

Chia Oil Microencapsulation Using Tannic Acid and Soy Protein Isolate as Wall Materials

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Oil Extraction and Preparation of Microcapsules
Quantification of Cross-Linker Consumption
2.3. Characterization of Microcapsules
2.3.1. Morphological Analysis
2.3.2. Contact Angle
2.3.3. Size Distribution of Oil Droplets in Reconstituted Emulsions
2.3.4. Color Measurements
2.3.5. Moisture Content and Water Activity
2.3.6. Determination of Encapsulation Efficiency
2.4. Oil Oxidative Stability Study
2.5. Fatty Acid Composition
2.6. Storage Test
2.7. In Vitro Gastric-Intestinal Digestion
2.7.1. Total Polyphenol Content
2.7.2. In Vitro Antioxidant Capacity
Reducing Power
Radical Scavenging Activity
2.8. Statistical Analysis
3. Results and Discussion
3.1. Quantification of Cross-Linker Consumption
3.2. Characterization of Microcapsules
3.3. Oxidative Stability of Microcapsules
3.4. Storage Test
3.5. In Vitro Gastrointestinal Digestion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sosa, M.D.; Magallanes, L.M.; Grosso, N.R.; Pramparo, M.D.C.; Gayol, M.F. Optimisation of omega-3 concentration and sensory analysis of chia oil. Ind. Crops Prod. 2020, 154, 112635. [Google Scholar] [CrossRef]
- Racine, R.A.; Deckelbaum, R.J. Sources of the very-long-chain unsaturated omega-3 fatty acids: Eicosapentaenoic acid and docosahexaenoic acid. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 123–128. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Martínez, M.L.; Paredes, A.J.; León, A.E.; Ribotta, P.D. Study of the preparation process and variation of wall components in chia (Salvia hispanica L.) oil microencapsulation. Powder Technol. 2016, 301, 868–875. [Google Scholar] [CrossRef]
- Sundar, S.K.; Parikh, J.K. Advances and trends in encapsulation of essential oils. Int. J. Pharm. 2023, 635, 122668. [Google Scholar] [CrossRef] [PubMed]
- Heck, R.T.; Lorenzo, J.M.; Dos Santos, B.A.; Cichoski, A.J.; de Menezes, C.R.; Campagnol, P.C.B. Microencapsulation of healthier oils: An efficient strategy to improve the lipid profile of meat products. Curr. Opin. Food Sci. 2020, 40, 6–12. [Google Scholar] [CrossRef]
- González, A.; Strumia, M.C.; Alvarez Igarzabal, C.I. Cross-linked soy protein as material for biodegradable films: Synthesis, characterization and biodegradation. J. Food Eng. 2011, 106, 331–338. [Google Scholar] [CrossRef]
- Picchio, M.L.; Linck, Y.G.; Monti, G.A.; Gugliotta, L.M.; Minari, R.J.; Alvarez Igarzabal, C.I. Casein films crosslinked by tannic acid for food packaging applications. Food Hydrocoll. 2018, 84, 424–434. [Google Scholar] [CrossRef]
- Muhoza, B.; Xia, S.; Zhang, X. Gelatin and high methyl pectin coacervates crosslinked with tannic acid: The characterization, rheological properties, and application for peppermint oil microencapsulation. Food Hydrocoll. 2019, 97, 105174. [Google Scholar] [CrossRef]
- Shavandi, A.; Bekhit, A.E.D.A.; Saeedi, P.; Izadifar, Z.; Bekhit, A.A.; Khademhosseini, A. Polyphenol uses in biomaterials engineering. Biomaterials 2018, 167, 91–106. [Google Scholar] [CrossRef]
- Guo, Y.; Bao, Y.H.; Sun, K.F.; Chang, C.; Liu, W.F. Effects of covalent interactions and gel characteristics on soy protein-tannic acid conjugates prepared under alkaline conditions. Food Hydrocoll. 2021, 112, 106293. [Google Scholar] [CrossRef]
- Kim, T.J.; Silva, J.L.; Jung, Y.S. Enhanced functional properties of tannic acid after thermal hydrolysis. Food Chem. 2011, 126, 116–120. [Google Scholar] [CrossRef]
- Kim, T.J.; Silva, J.L.; Kim, M.K.; Jung, Y.S. Enhanced antioxidant capacity and antimicrobial activity of tannic acid by thermal processing. Food Chem. 2010, 118, 740–746. [Google Scholar] [CrossRef]
- Martínez, M.L.; Marín, M.A.; Salgado Faller, C.M.; Revol, J.; Penci, M.C.; Ribotta, P.D. Chia (Salvia hispanica L.) oil extraction: Study of processing parameters. LWT—Food Sci. Technol. 2012, 47, 78–82. [Google Scholar] [CrossRef]
- González, A.; Martínez, M.L.; León, A.E.; Ribotta, P.D. Effects on bread and oil quality after functionalization with microencapsulated chia oil. J. Sci. Food Agric. 2018, 98, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Rouquerol, J.; Baron, G.; Denoyel, R.; Giesche, H.; Groen, J.; Klobes, P.; Levitz, P.; Neimark, A.V.; Rigby, S.; Skudas, R. Recommendations for the characterization of porous solids (Technical Report). Pure Appl. Chem. 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Romero, M.R.; Wolfel, A.; Igarzabal, C.I.A. Smart valve: Polymer actuator to moisture soil control. Sens. Actuators B Chem. 2016, 234, 53–62. [Google Scholar] [CrossRef]
- Bordón, M.G.; Alasino, N.P.X.; Martínez, V.; Gauna Peter, R.; Iturralde, R.; Ribotta, P.D.; Martínez, M.L. Influence of the spray drying operating conditions on the estimated drying kinetics of emulsion single droplets and the properties of microencapsulated chia oil. Powder Technol. 2021, 383, 302–317. [Google Scholar] [CrossRef]
- AOCS. Official Methods and Recommended Practices of the American Oil Chemists’ Society, 5th ed.; AOCS Press: Champaign, IL, USA, 2009. [Google Scholar]
- Ganañ, N.; Bordón, M.G.; Ribotta, P.D.; González, A. Study of chia oil microencapsulation in soy protein microparticles using supercritical Co2-assisted impregnation. J. CO2 Util. 2020, 40, 101221. [Google Scholar] [CrossRef]
- Lucini Mas, A.; Brigante, F.I.; Salvucci, E.; Pigni, N.B.; Martinez, M.L.; Ribotta, P.; Wunderlin, D.A.; Baroni, M.V. Defatted chia flour as functional ingredient in sweet cookies. How do Processing, simulated gastrointestinal digestion and colonic fermentation affect its antioxidant properties? Food Chem. 2020, 316, 126279. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A. Colorimetry of total phenolics with phosphomo-lybdic-phosphotungstic acid reagents. Am. J. Enol. Vitic. 1965, 16, 144–158. [Google Scholar] [CrossRef]
- Benzie, F.F.; Strainb, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Apllying an Improved ABTS Radical Cation Decolorization Assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT—Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Goyal, A.; Sharma, V.; Sihag, M.K.; Tomar, S.K.; Arora, S.; Sabikhi, L.; Singh, A.K. Development and physico-chemical characterization of microencapsulated flaxseed oil powder: A functional ingredient for omega-3 fortification. Powder Technol. 2015, 286, 527–537. [Google Scholar] [CrossRef]
- Copado, C.N.; Julio, L.M.; Diehl, B.W.K.; Ixtaina, V.Y.; Tomás, M.C. Multilayer microencapsulation of chia seed oil by spray-drying using electrostatic deposition technology. LWT 2021, 152, 112206. [Google Scholar] [CrossRef]
- Bordón, M.G.; Bodoira, R.M.; González, A.; Piloni, R.; Ribotta, P.D.; Martínez, M.L. Spray-Drying, Oil Blending, and the Addition of Antioxidants Enhance the Storage Stability at Room Temperature of Omega-3-Rich Microcapsules Based on Chia Oil. Eur. J. Lipid Sci. Technol. 2022, 124, 2100181. [Google Scholar] [CrossRef]
- Di Giorgio, L.; Salgado, P.R.; Mauri, A.N. Encapsulation of fish oil in soybean protein particles by emulsification and spray drying. Food Hydrocoll. 2019, 87, 891–901. [Google Scholar] [CrossRef]
- Timilsena, Y.P.; Adhikari, R.; Barrow, C.J.; Adhikari, B. Microencapsulation of chia seed oil using chia seed protein isolate-chia seed gum complex coacervates. Int. J. Biol. Macromol. 2016, 91, 347–357. [Google Scholar] [CrossRef]
- Copado, C.N.; Diehl, B.W.K.; Ixtaina, V.Y.; Tomás, M.C. Application of Maillard reaction products on chia seed oil microcapsules with different core/wall ratios. LWT 2017, 86, 408–417. [Google Scholar] [CrossRef]
- Alimentarius, C. Fats, Oils and Related Products; FAO: Rome, Italy, 2001. [Google Scholar]
- Martínez, M.L.; Curti, M.I.; Roccia, P.; Llabot, J.M.; Penci, M.C.; Bodoira, R.M.; Ribotta, P.D. Oxidative stability of walnut (Juglans regia L.) and chia (Salvia hispanica L.) oils microencapsulated by spray drying. Powder Technol. 2015, 270, 271–277. [Google Scholar] [CrossRef]
- Ixtaina, V.Y.; Julio, L.M.; Wagner, J.R.; Nolasco, S.M.; Tomás, M.C. Physicochemical characterization and stability of chia oil microencapsulated with sodium caseinate and lactose by spray-drying. Powder Technol. 2015, 271, 26–34. [Google Scholar] [CrossRef]
- Locali Pereira, A.R.; Gonçalves Cattelan, M.; Nicoletti, V.R. Microencapsulation of pink pepper essential oil: Properties of spray-dried pectin/SPI double-layer versus SPI single-layer stabilized emulsions. Colloids Surf. A Physicochem. Eng. Asp. 2019, 581, 123806. [Google Scholar] [CrossRef]
- Jakobek, L. Interactions of polyphenols with carbohydrates, lipids and proteins. Food Chem. 2015, 175, 556–567. [Google Scholar] [CrossRef]
- Inapurapu, S.P.; Ibrahim, A.; Kona, S.R.; Pawar, S.C.; Bodiga, S.; Bodiga, V.L. Development and characterization of ω-3 fatty acid nanoemulsions with improved physicochemical stability and bioaccessibility. Colloids Surf. A Physicochem. Eng. Asp. 2020, 606, 125515. [Google Scholar] [CrossRef]
- Li, R.; Dai, T.; Tan, Y.; Fu, G.; Wan, Y.; Liu, C.; McClements, D.J. Fabrication of pea protein-tannic acid complexes: Impact on formation, stability, and digestion of flaxseed oil emulsions. Food Chem. 2020, 310, 125828. [Google Scholar] [CrossRef] [PubMed]
- Everette, J.D.; Bryant, Q.M.; Green, A.M.; Abbey, Y.A.; Wangila, G.W.; Walker, R.B. Thorough study of reactivity of various compound classes toward the folin-Ciocalteu reagent. J. Agric. Food Chem. 2010, 58, 8139–8144. [Google Scholar] [CrossRef]
- Brune, M.; Rossander, L.; Hallberg, L. Iron Absorp-tion and Phenolic Compounds: Importance of Different Phenolic Structures. Eur. J. Clin. Nutr. 1989, 43, 547–558. [Google Scholar]
- Badhani, B.; Sharma, N.; Kakkar, R. Gallic acid: A versatile antioxidant with promising therapeutic and industrial applications. RSC Adv. 2015, 5, 27540–27557. [Google Scholar] [CrossRef]
- Yen, G.-C.; Duh, P.-D.; Tsai, H.-L. Antioxidant and pro-oxidant properties of ascorbic acid and gallic acid. Food Chem. 2002, 79, 307–313. [Google Scholar] [CrossRef]
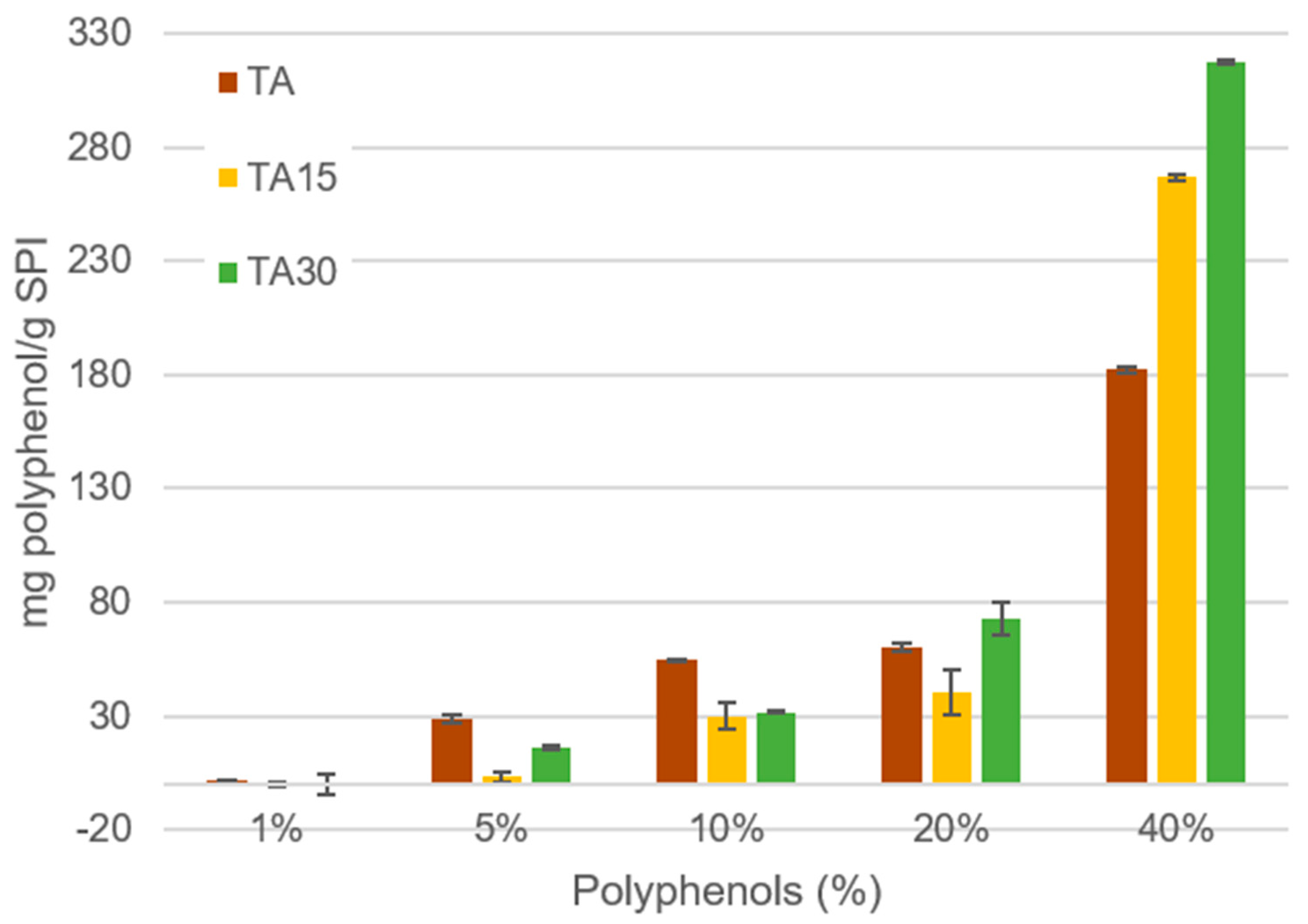
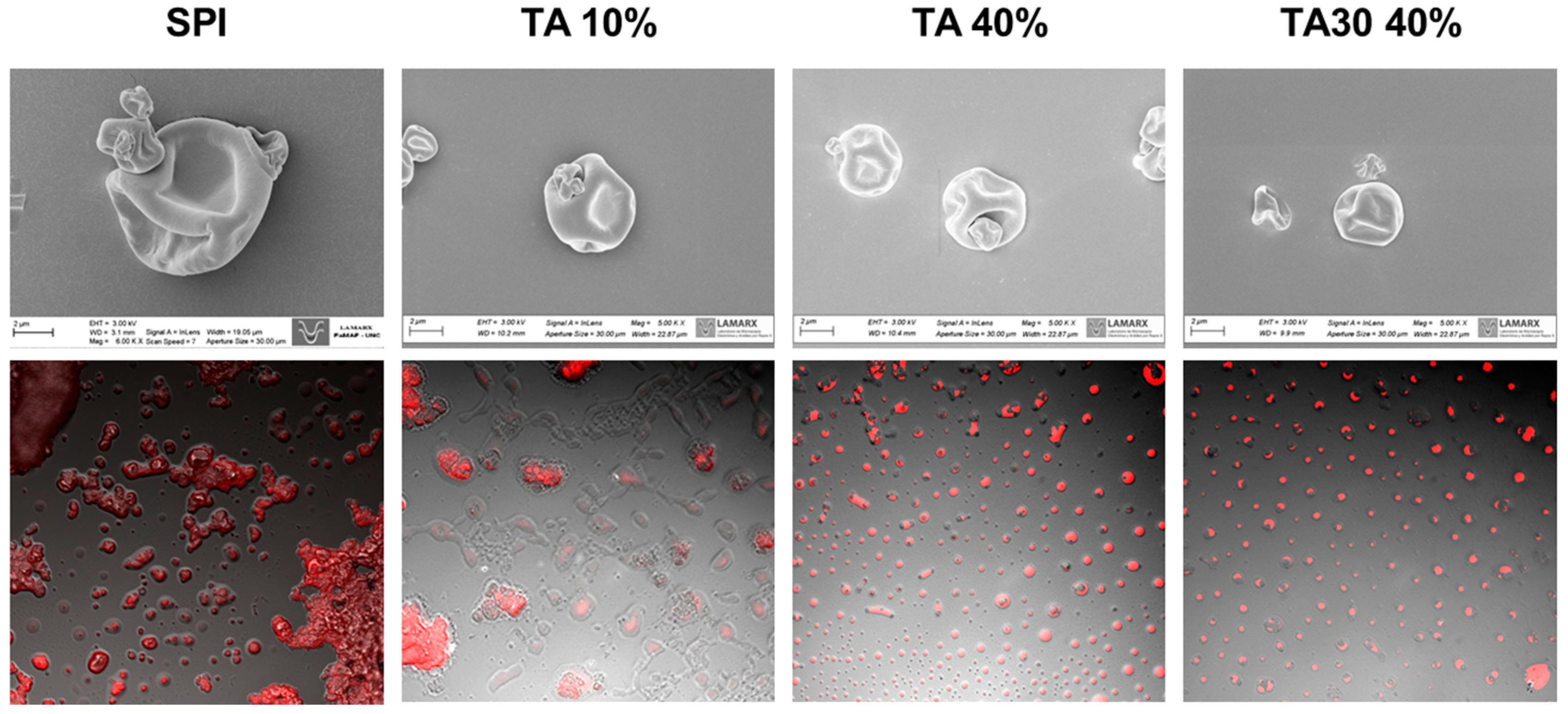


{kind=link}
{kind=link}
{kind=link}
| Samples | L* | a* | b* | WI | YI |
|---|---|---|---|---|---|
| SPI | 89.46 ± 0.04 L | −0.05 ± 0.01 A | 10.97 ± 0.04 A | 56.54 ± 0.09 H | 17.52 ± 0.06 A |
| TA 1% | 82.19 ± 0.25 K | 1.22 ± 0.10 E | 16.16 ± 0.31 B | 33.72 ± 1.25 G | 28.08 ± 0.61 B |
| TA15 1% | 81.39 ± 0.38 J | 1.99 ± 0.08 H | 16.10 ± 0.18 B | 33.08 ± 0.91 G | 28.27 ± 0.44 B |
| TA30 1% | 81.31 ± 0.38 J | 2.01 ± 0.11 H | 15.64 ± 0.27 B | 34.40 ± 1.10 G | 27.48 ± 0.57 B |
| TA 5% | 68.27 ± 0.55 H | 1.89 ± 0.10 G | 25.51 ± 0.15 H | −8.27 ±0.70 B | 53.39 ± 0.53 G |
| TA15 5% | 67.27 ± 0.09 G | 0.36 ± 0.01 B | 20.08 ± 0.01 E | 7.03 ± 0.07 F | 42.65 ± 0.04 C |
| TA30 5% | 70.60 ± 0.45 I | 2.69 ± 0.10 I | 25.17 ± 0.07 H | −4.90 ± 0.43 C | 50.93 ± 0.31 F |
| TA 10% | 60.32 ± 0.80 F | −0.10 ± 0.03 A | 20.07 ± 0.22 E | 0.11 ± 0.71 D | 47.53 ± 0.56 E |
| TA15 10% | 57.32 ± 0.10 D | 6.09 ± 0.01 J | 25.78 ± 0.02 H | −20.01 ± 0.07 A | 64.25 ± 0.08 J |
| TA30 10% | 60.59 ± 0.15 F | 1.01 ± 0.00 D | 22.10 ± 0.06 G | −5.72 ± 0.02 C | 52.11 ± 0.01 G |
| TA20% | 55.62 ± 0.34 C | −0.13 ± 0.18 A | 21.25 ± 0.88 F | −8.13 ± 2.82 B | 54.59 ± 2.45 H |
| TA15 20% | 53.72 ± 0.48 B | 0.81 ± 0.06 C | 19.76 ± 0.12 E | −5.56 ± 0.16 C | 52.55 ± 0.18 G |
| TA30 20% | 59.65 ± 0.10 E | −0.16 ± 0.02 A | 17.96 ± 0.03 C | 5.77 ± 0.07 F | 43.01 ± 0.05 C |
| TA 40% | 44.45 ± 0.36 A | 1.13 ± 0.01 E | 17.67 ± 0.39 C | −8.55 ± 0.84 B | 56.78 ± 0.83 I |
| TA15 40% | 60.55 ± 0.05 F | 1.70 ± 0.01 F | 19.09 ± 0.02 D | 3.27 ± 0.08 E | 45.05 ± 0.06 D |
| TA30 40% | 55.15 ± 0.32 C | 2.10 ± 0.01 H | 18.92 ± 0.10 D | −1.61 ± 0.63 D | 49.01 ± 0.55 E |
| Samples | MC (%) | EE (%) | IP (h) | PF | D[4,3] | PDI |
|---|---|---|---|---|---|---|
| Bulk oil | - | - | 2.51 ± 0.15 A | - | - | - |
| SPI | 3.16 ± 0.18 A | 60.21 ± 1.06 A | 5.35 ± 0.12 B | 2.13 | 10.52 ± 0.46 B | 0.89 ± 0.04 A |
| TA 1% | 4.14 ± 0.02 B | 75.18 ± 17.84 A | 7.56 ± 0.16 D | 3.01 | 1.37 ± 0.00 A | 0.93 ± 0.00 A |
| TA15 1% | 3.16 ± 0.05 A | 70.38 ± 4.28 A | 7.94 ± 0.01 D | 3.16 | 2.54 ± 2.08 A | 1.06 ± 0.28 A |
| TA30 1% | 3.24 ± 0.43 A | 69.32 ± 1.38 A | 7.26 ± 0,10 D | 2.89 | 1.57 ± 0.02 A | 0.95 ± 0.00 A |
| TA 5% | 4.59 ± 0.25 C | 73.29 ± 8.05 A | 11.97 ± 0.63 F | 4.77 | 1.34 ± 0.00 A | 0.91 ± 0.01 A |
| TA15 5% | 3.26 ± 0.16 A | 65.99 ± 7.67 A | 7.62 ± 0.05 D | 3.04 | 1.59 ± 0.00 A | 0.95 ± 0.00 A |
| TA30 5% | 5.86 ± 0.13 D | 76.95 ± 9.19 A | 11.87 ± 0.54 F | 4.73 | 1.39 ± 0.00 A | 0.86 ± 0.00 A |
| TA 10% | 3.76 ± 0.44 B | 70.07 ± 3.50 A | 11.00 ± 0.25 E | 4.38 | 2.18 ± 0.23 A | 1.04 ± 0.05 A |
| TA15 10% | 3.82 ± 0.13 B | 54.62 ± 3.75 A | 6.63 ± 0.06 C | 2.64 | 12.42 ± 0.59 B | 1.04 ± 0.01 A |
| TA30 10% | 3.57 ± 0.44 B | 67.43 ± 9.12 A | 10.90 ± 0.27 E | 4.34 | 13.27 ± 0.72 B | 0.97 ± 0.06 A |
| TA 20% | 5.58 ± 0.10 D | 61.51 ± 4.97 A | 15.10 ± 0.19 G | 6.01 | 21.84 ± 2.45 C | 1.18 ± 0.06 A |
| TA15 20% | 3.67 ± 0.05 B | 78.38 ± 8.49 A | 15.87 ± 0.16 H | 6.32 | 18.10 ± 0.36 C | 1.03 ± 0.01 A |
| TA30 20% | 4.06 ± 0.05 B | 68.37 ± 0.57 A | 15.95 ± 0.25 H | 6.35 | 2.92 ± 1.55 A | 1.12 ± 0.20 A |
| TA 40% | 4.74 ± 0.24 C | 59.93 ± 0.37 A | 21.48 ± 0.28 I | 8.55 | 0.82 ± 0.02 A | 0.87 ± 0.03 A |
| TA15 40% | 3.43 ± 0.16 A | 76.28 ±13.77 A | 27.94 ± 0.88 J | 11.12 | 1.74 ± 0.39 A | 0.97 ± 0.04 A |
| TA30 40% | 3.29 ± 0.01 A | 63.76 ± 4.01 A | 22.17 ± 0.09 I | 8.83 | 1.51 ± 0.06 A | 0.95 ± 0.01 A |
| Samples | TPA [m2/g] | % Porosity |
|---|---|---|
| SPI | 22.10 A | 42.15 A |
| TA 40% | 17.75 A | 40.50 A |
| TA15 40% | 17.10 A | 40.00 A |
| Samples | Days | Saturated | C18:1 | C18:2 | C18:3 | MOC (%) | aw | D[4,3] | PDI |
|---|---|---|---|---|---|---|---|---|---|
| SPI | 0 | 96.04 ± 2.19 Aa | 64.18 ± 0.42 Aa | 191.47 ± 0.96 Aa | 649.20 ± 3.03 Ba | 2.16 ± 0.02 Aa | 0.168 ± 0.001 Aa | 12.27 ± 0.04 Ad | 1.10 ± 0.00 Aa |
| 180 | 230.16 ± 2.39 Bc | 168.34 ± 2.21 Bb | 228.65 ± 2.08 Bc | 333.22 ± 1.54 Aa | 3.29 ± 0.13 Ba | 0.285 ± 0.008 Ba | 217.27 ± 42.86 Bb | 2.42 ± 0.09 Ba | |
| TA 5% | 0 | 94.38 ± 0.27 Aa | 64.72 ± 0.42 Aa | 194.50 ± 0.43 Ba | 645.49 ± 3.02 Ba | 3.45 ± 0.35 Ab | 0.170 ± 0.001 Aa | 2.53 ± 0.08 Ac | 4.38 ± 0.17 Bc |
| 180 | 97.95 ± 0.39 Bb | 65.42 ± 0.53 Aa | 192.55 ± 0.40 Ab | 636.14 ± 0.40 Ab | 3.45 ± 0.35 Aa | 0.206 ± 0.008 Ab | 15.55 ± 0.25 Ba | 1.21 ± 0.08 Aa | |
| TA15 5% | 0 | 98.61 ± 0.08 Ba | 65.50 ± 0.65 Ba | 193.70 ± 0.43 Aa | 628.38 ± 3.65 Aa | 3.27 ± 0.47 Ab | 0.178 ± 0.002 Aa | 2.61 ± 0.03 Ac | 5.32 ± 0.37 Bd |
| 180 | 93.96 ± 1.08 Aa | 63.22 ± 0.27 Aa | 194.04 ± 0.67 Ab | 646.38 ± 4.88 Bc | 3.67 ± 0.40 Aa | 0.227 ± 0.008 Bc | 18.81 ± 1.30 Ba | 0.95 ± 0.06 Aa | |
| TA 40% | 0 | 98.49 ± 0.08 Ba | 66.42 ± 0.65 Aa | 194.39 ± 0.44 Ba | 642.11 ± 3.02 Aa | 2.28 ± 0.13 Aa | 0.157 ± 0.002 Aa | 2.24 ± 0.02 Aa | 3.58 ± 0.27 Ab |
| 180 | 90.44 ± 2.35 Aa | 61.07 ± 1.92 Aa | 186.96 ± 1.05 Aa | 655.79 ± 3.44 Bd | 3.28 ± 0.14 Ba | 0.163 ± 0.003 Bd | 22.07 ± 3.82 Ba | 1.02 ± 0.08 Aa | |
| TA15 40% | 0 | 94.00 ± 0.27 Ba | 63.90 ± 0.42 Ba | 192.84 ± 0.97 Ba | 647.41 ± 3.03 Aa | 2.83 ± 0.16 Ab | 0.175 ± 0.002 Ba | 2.37 ± 0.02 Ab | 6.03 ± 0.06 Be |
| 180 | 92.16 ± 1.02 Aa | 61.30 ± 1.87 Aa | 187.47 ± 1.04 Aa | 655.49 ± 3.42 Bd | 3.39 ± 0.18 Ba | 0.168 ± 0.001 Ad | 19.33 ± 0.33 Ba | 0.95 ± 0.03 Aa |
| Sample | % OA | Saturated (mg/g Oil) | C18:1 (mg/g Oil) | C18:2 (mg/g Oil) | C18:3 (mg/g Oil) |
|---|---|---|---|---|---|
| Bulk oil | 100.00 ± 0.06 D | 101.99 ± 25.01 A | 74.62 ± 6.53 A | 209.51 ± 26.04 A | 613.89 ± 58.08 A |
| SPI | 95.17 ± 0.46 C | 99.49 ± 4.45 A | 66.43 ± 3.37 A | 202.80 ± 1.71 A | 612.44 ± 31.11 A |
| TA 40% | 48.45 ± 0.30 B | 105.87 ± 8.22 A | 81.39 ± 0.42 A | 192.69 ± 18.31 A | 638.51 ± 39.52 A |
| TA15 40% | 35.94 ± 2.73 A | 89.04 ± 23.35 A | 71.64 ± 4.36 A | 204.48 ± 4.44 A | 632.17 ± 22.17 A |
| Sample | TPC (mg GAE/g) | FRAP (mg TE/g) | TEAC (mg TE/g) | DPPH (mg TE/g) |
|---|---|---|---|---|
| Undigested | ||||
| SPI | 23.40 ± 9.40 b* | <LOD | <LOD | <LOD |
| TA 40% | 238.10 ± 13.83 a* | 554.22 ± 20.91 a | 594.89 ± 16.61 a* | 528.88 ± 33.40 ab* |
| TA15 40% | 227.07 ± 19.23 a* | 569.74 ± 30.85 a* | 625.75 ± 34.56 ab* | 469.63 ± 86.11 b* |
| Digested | ||||
| SPI | 46.62 ± 6.59 C* | <LOD | <LOD | <LOD |
| TA 40% | 322.09 ± 10.08 B* | 663.61 ± 132.02 AB | 917.25 ± 61.69 AB* | 818.92 ± 64.94 AB* |
| TA15 40% | 382.35 ± 95.43 A* | 805.82 ± 118.75 A* | 1035.17 ± 232.23 A* | 916.01 ± 180.35 A* |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimenez, P.A.; Lucini Mas, A.; Ribotta, P.D.; Martínez, M.L.; González, A. Chia Oil Microencapsulation Using Tannic Acid and Soy Protein Isolate as Wall Materials. Foods 2023, 12, 3833. https://doi.org/10.3390/foods12203833
Gimenez PA, Lucini Mas A, Ribotta PD, Martínez ML, González A. Chia Oil Microencapsulation Using Tannic Acid and Soy Protein Isolate as Wall Materials. Foods. 2023; 12(20):3833. https://doi.org/10.3390/foods12203833
Chicago/Turabian StyleGimenez, Paola Alejandra, Agustín Lucini Mas, Pablo Daniel Ribotta, Marcela Lilian Martínez, and Agustín González. 2023. "Chia Oil Microencapsulation Using Tannic Acid and Soy Protein Isolate as Wall Materials" Foods 12, no. 20: 3833. https://doi.org/10.3390/foods12203833
APA StyleGimenez, P. A., Lucini Mas, A., Ribotta, P. D., Martínez, M. L., & González, A. (2023). Chia Oil Microencapsulation Using Tannic Acid and Soy Protein Isolate as Wall Materials. Foods, 12(20), 3833. https://doi.org/10.3390/foods12203833