
Characterizing the Contribution of Functional Microbiota Cultures in Pit Mud to the Metabolite Profiles of Fermented Grains

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of High Ability of PM Culture Producing Ethyl Hexanoate
2.2. Baijiu Brewing and Sampling
2.3. Physicochemical Properties Analyzing
2.4. Detecting Volatiles
2.5. Amplicon Sequencing
2.6. Metagenomic Sequencing
2.7. Statistical Analysis
3. Results and Discussion
3.1. Changes in Physicochemical Properties of FG
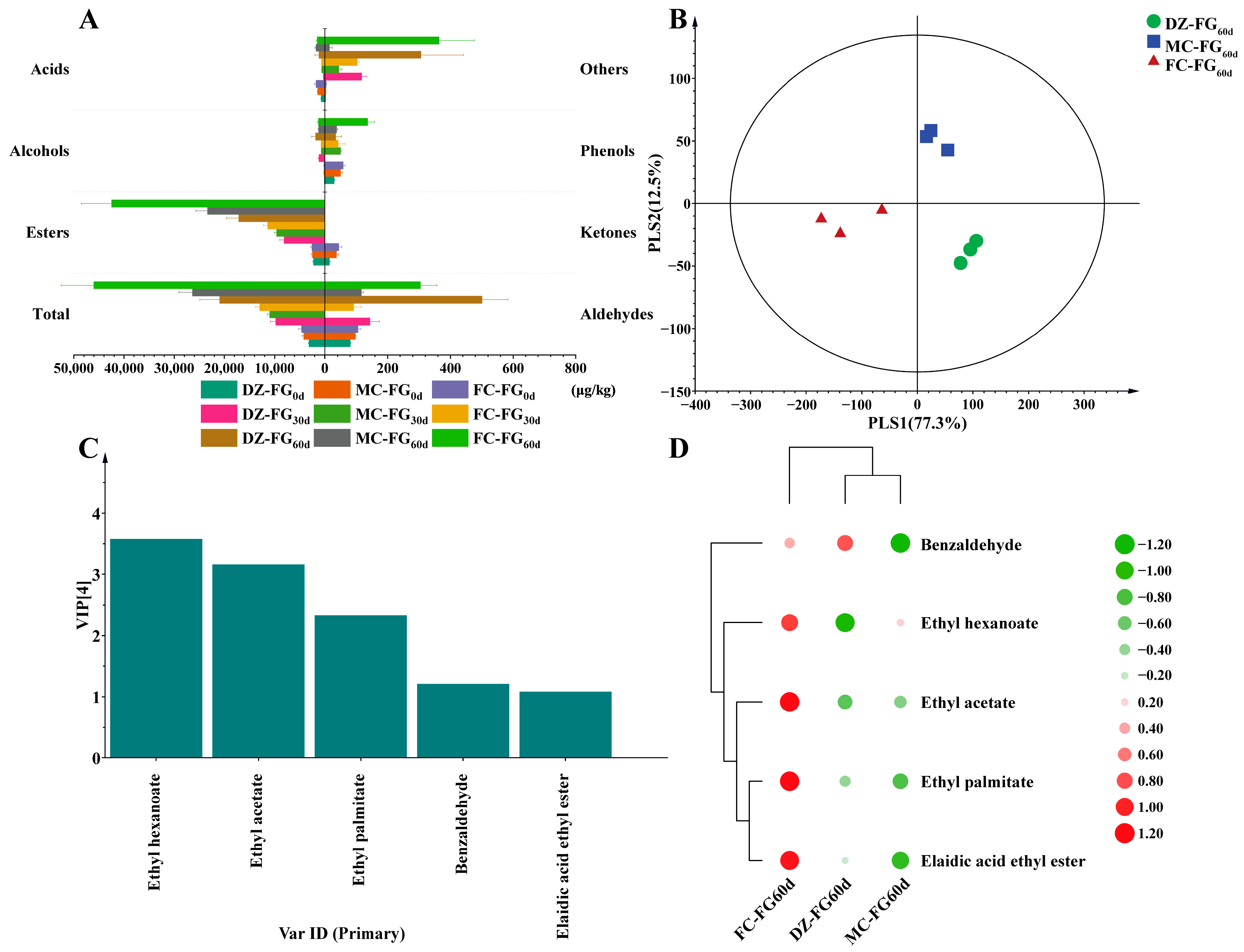
3.2. Changes in Volatile Components
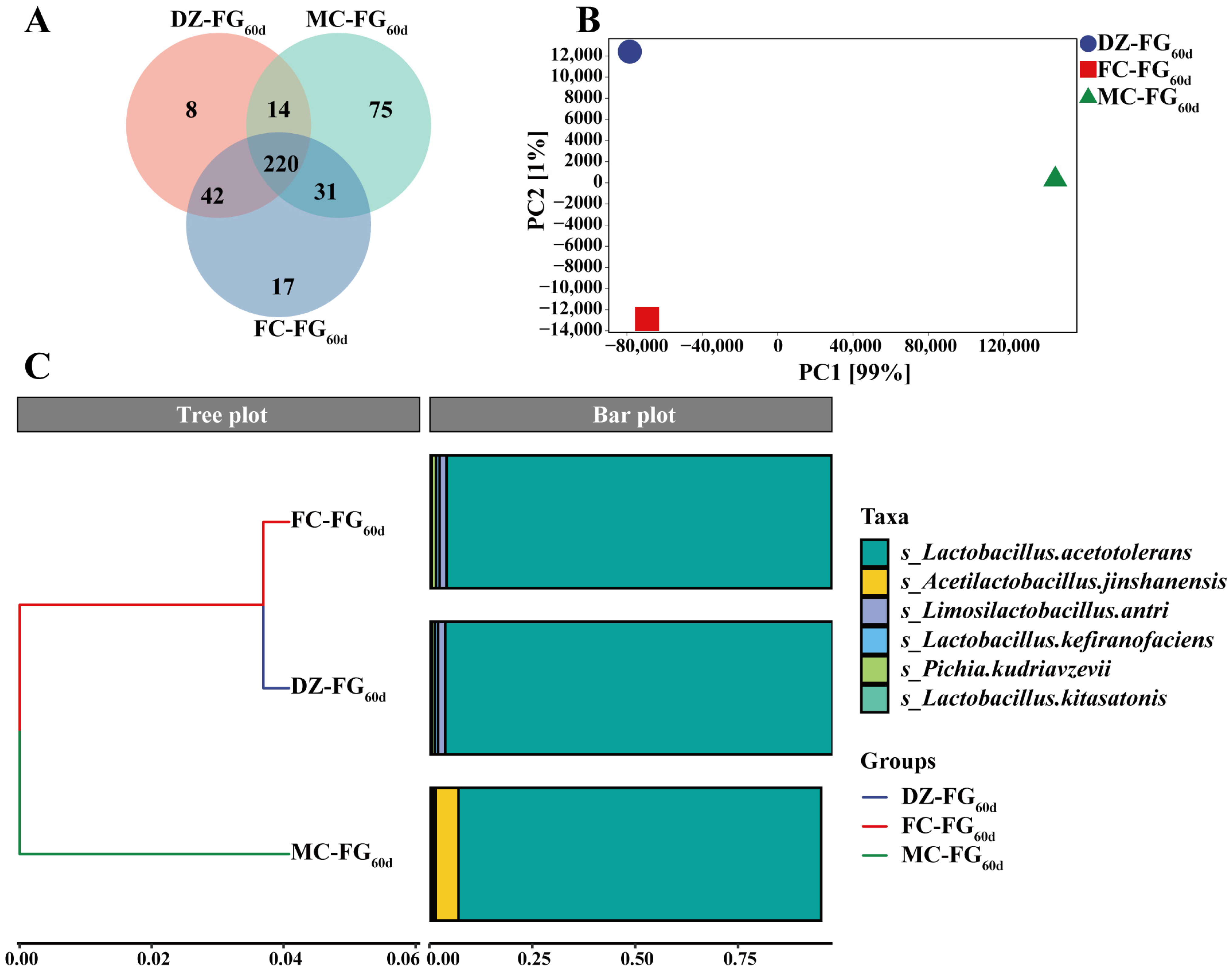
3.3. Difference in Microbiota Diversity and Their Constructure among Samples
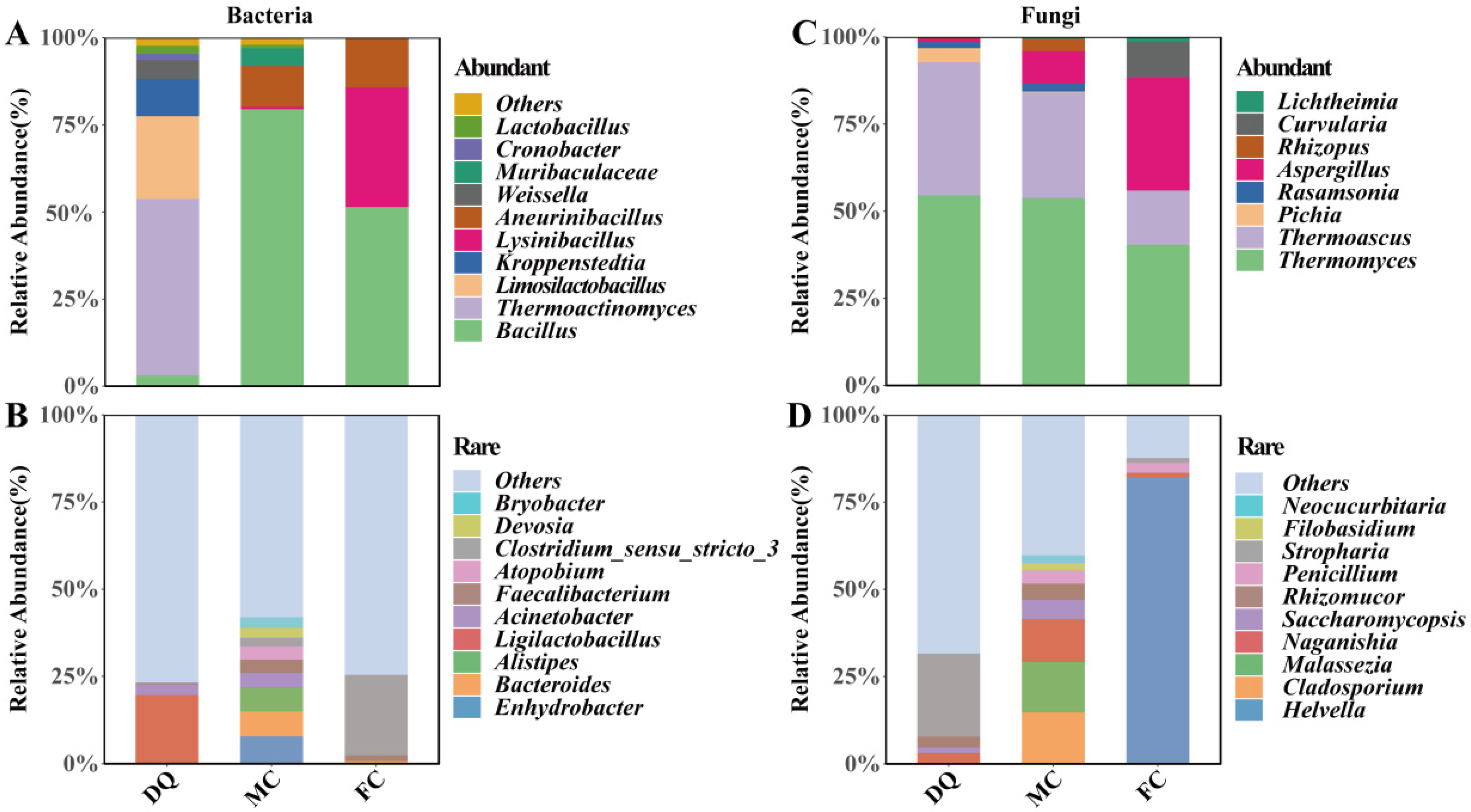
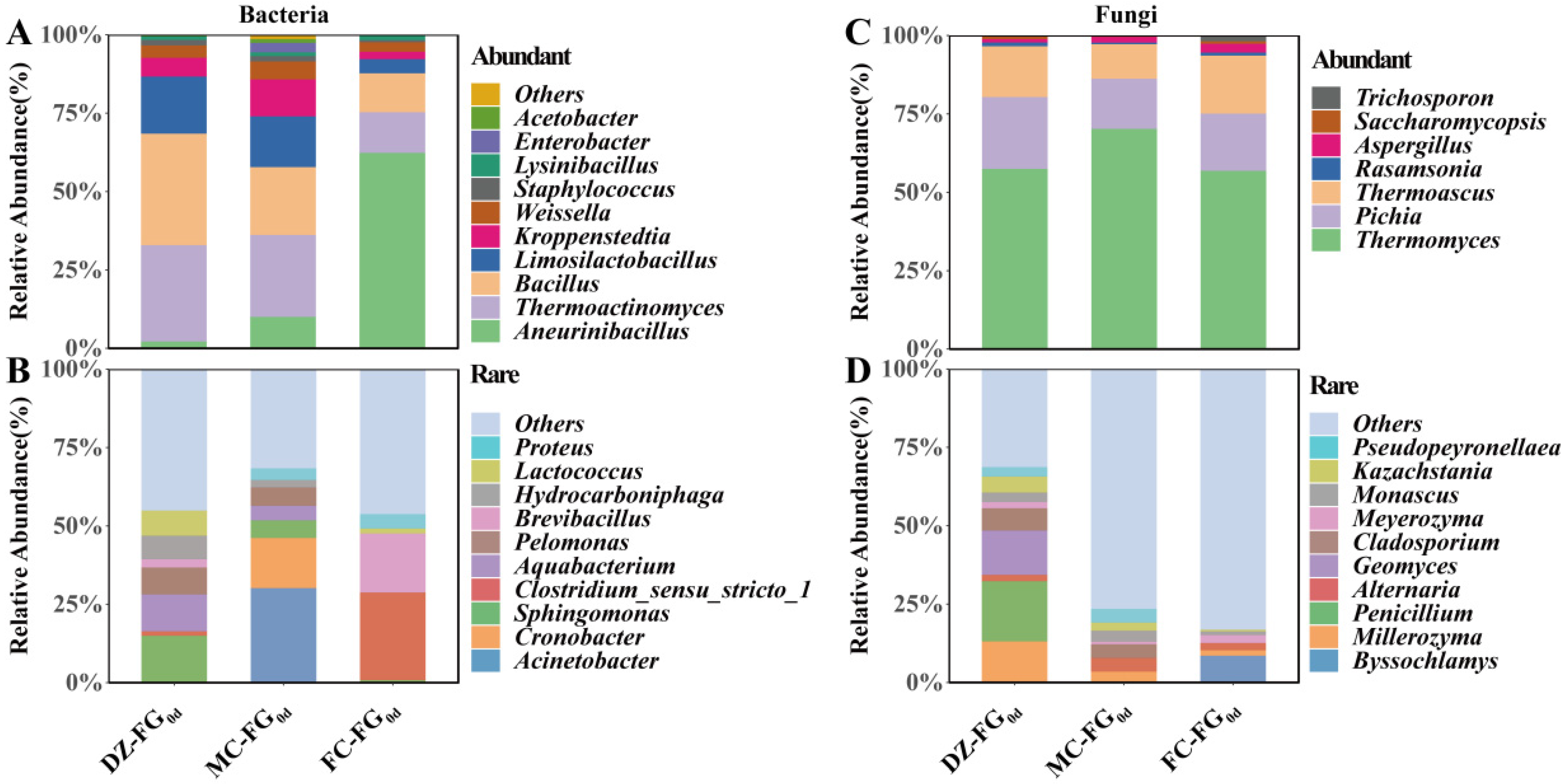
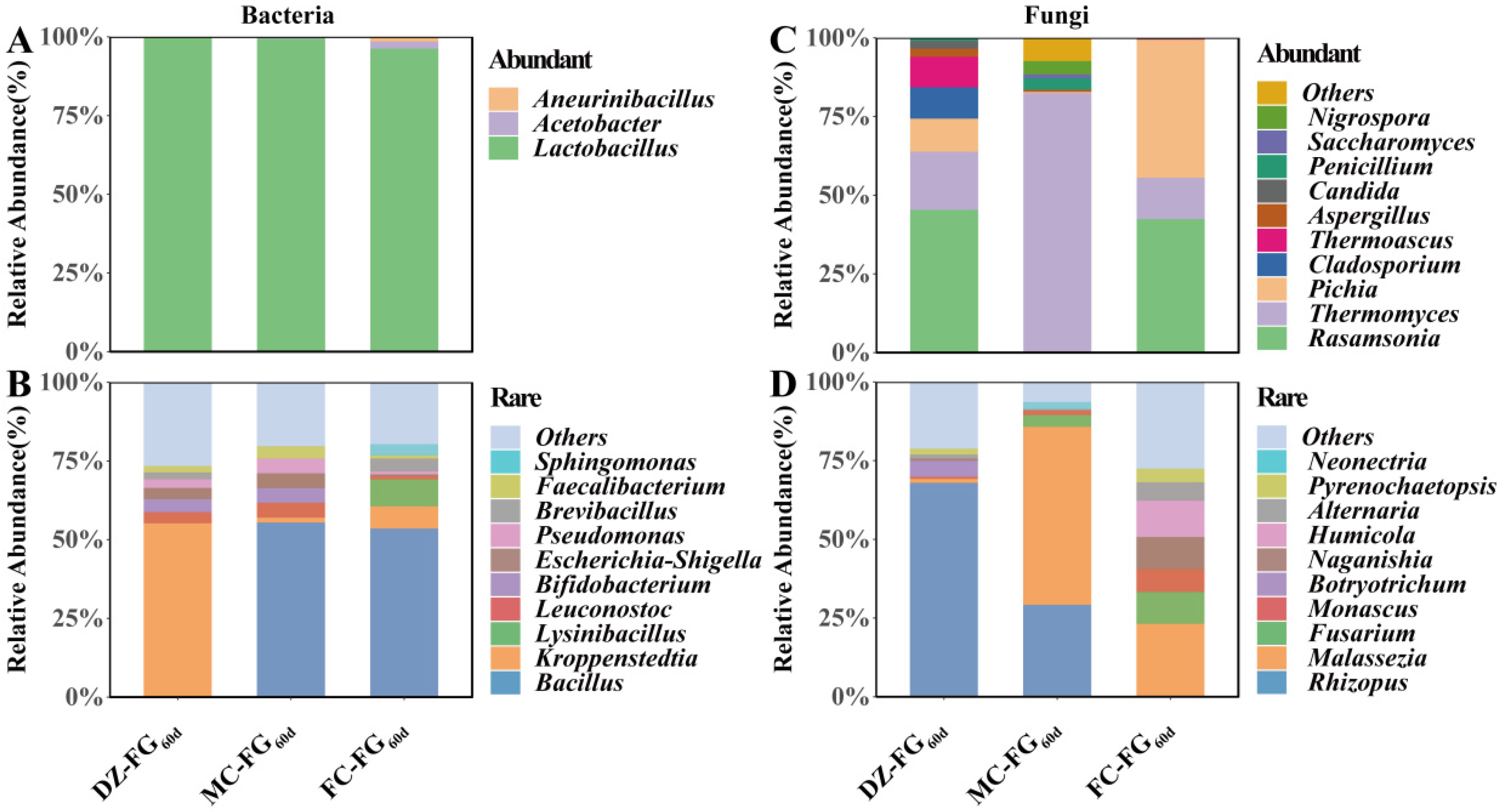
3.4. Distribution of Abundant and Rare Microbial Taxa
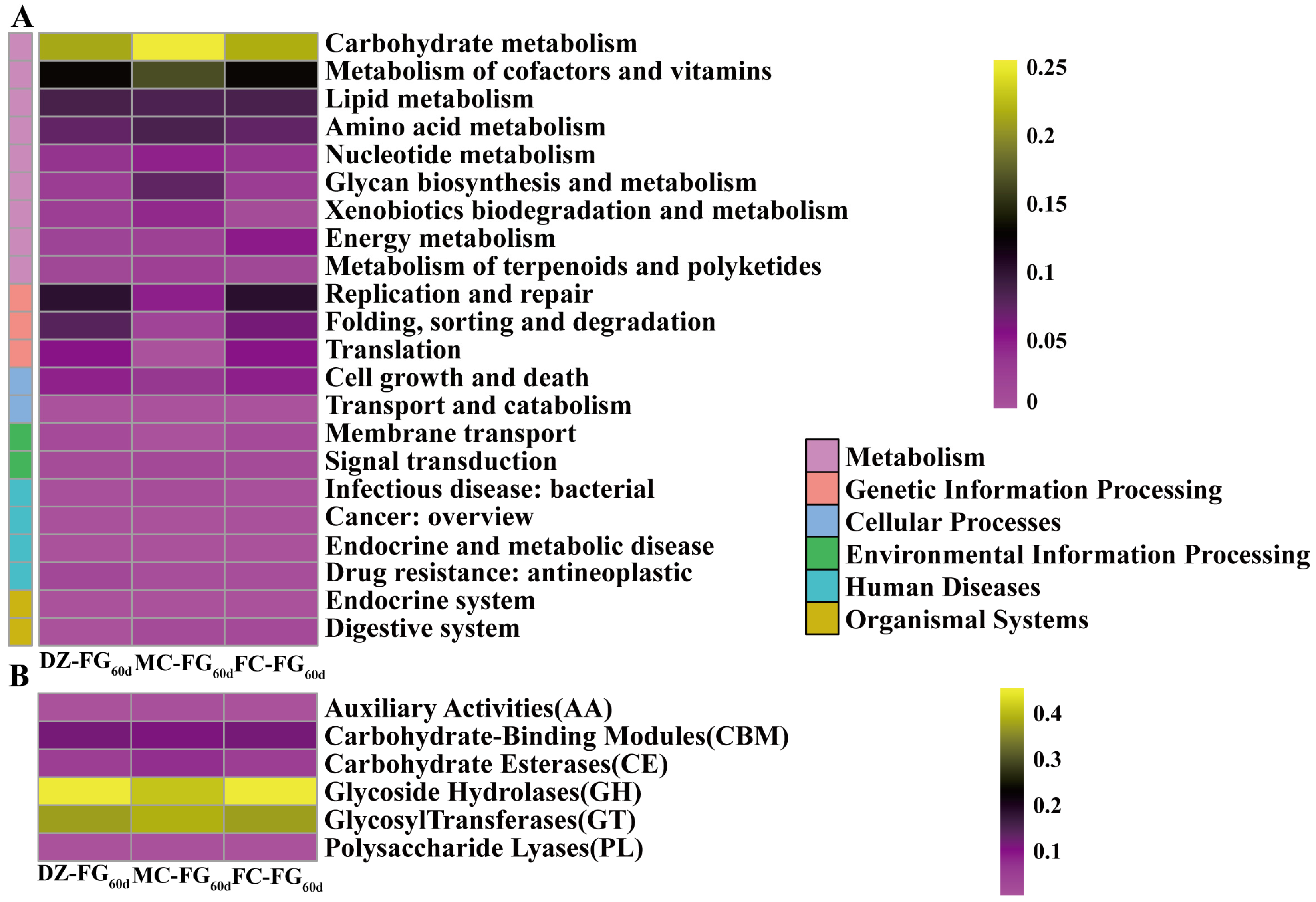
3.5. Differences in Microbiota Composition and Functional Annotation among FG by Metagenomic Sequencing
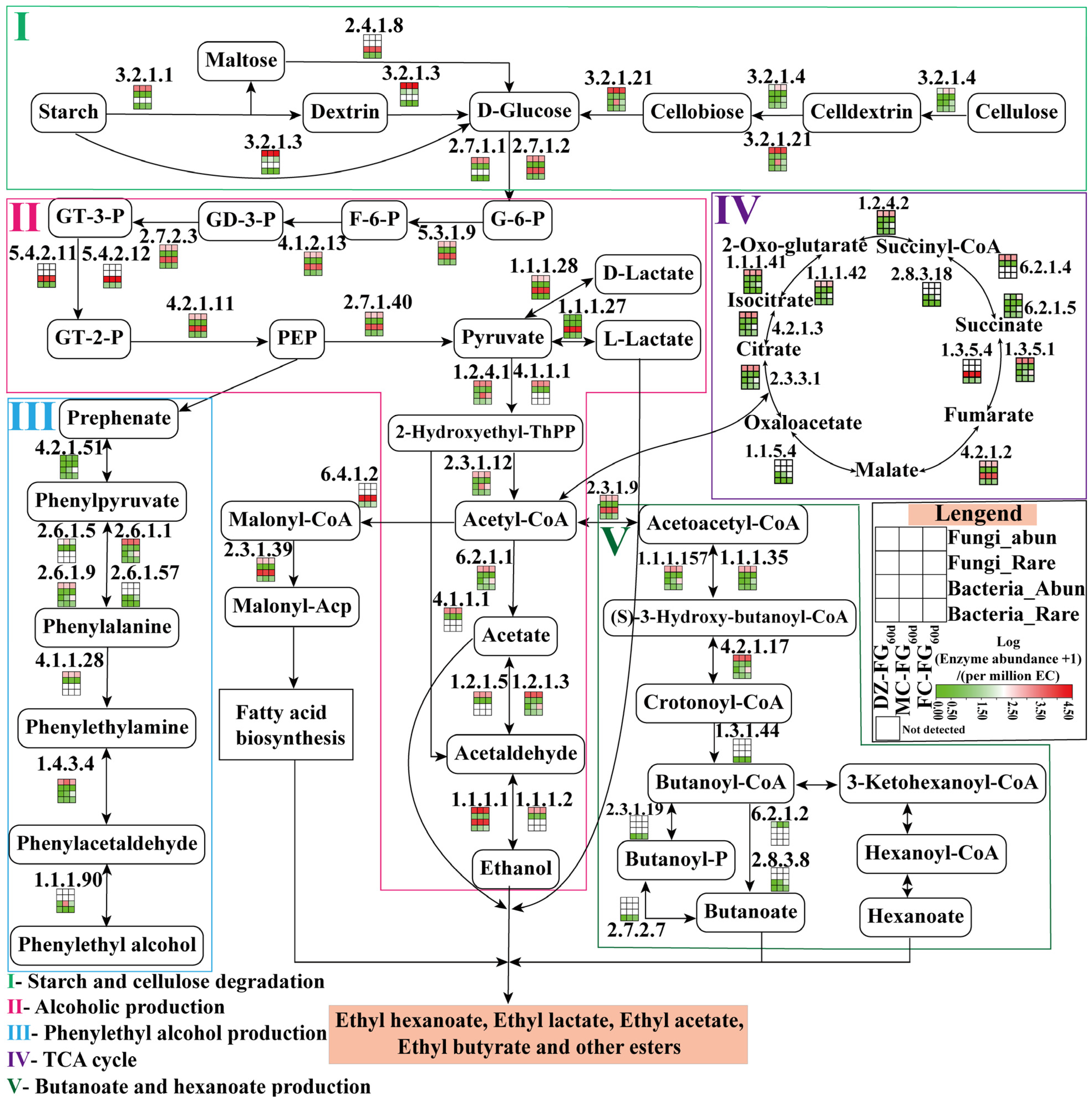
3.6. Differences in the Contribution of Abundant and Rare Taxa to Metabolic Pathways
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tao, Y.; Wang, X.; Li, X.; Wei, N.; Jin, H.; Xu, Z.; Tang, Q.; Zhu, X. The functional potential and active populations of the pit mud microbiome for the production of Chinese strong-flavour liquor. Microb. Biotechnol. 2017, 10, 1603–1615. [Google Scholar] [CrossRef]
- Gao, J.; Liu, G.; Li, A.; Liang, C.; Ren, C.; Xu, Y. Domination of pit mud microbes in the formation of diverse flavour compounds during Chinese strong aroma-type Baijiu fermentation. LWT 2021, 137, 110442. [Google Scholar] [CrossRef]
- Mu, Y.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H.; Dong, Y.; Wang, C.; Wang, X.; Pan, Q.; Tang, H. Comprehensive analysis for the bioturbation effect of space mutation and biofortification on strong-flavor Daqu by high-throughput sequencing, volatile analysis and metabolomics. Food Chem. 2023, 403, 134440. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H.; Tang, H.; Pan, Q.; Tang, H. Response and assembly of abundant and rare taxa in Zaopei under different combination patterns of Daqu and pit mud: From microbial ecology to Baijiu brewing microecosystem. Food Sci. Hum. Wellness 2024, 13, 1439–1452. [Google Scholar] [CrossRef]
- Qian, W.; Lu, Z.; Chai, L.; Zhang, X.; Li, Q.; Wang, S.; Shen, C.; Shi, J.; Xu, Z. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor Baijiu production. Food Res. Int. 2021, 147, 110449. [Google Scholar] [CrossRef]
- Mao, F.; Huang, J.; Zhou, R.; Zhang, S.; Qin, H. Effects of microbial community in artificial pit mud on the formation of flavor metabolites during the fermentation of Nong-flavor Baijiu. Food Sci. 2024, 45, 125–134. (In Chinese) [Google Scholar]
- Deng, L.; Mao, X.; Liu, D.; Ning, X.; Shen, Y.; Chen, B.; Nie, H.; Huang, D.; Luo, H. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu With Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, Y.; Guo, X.; Zhao, K.; Penttinen, P.; Tian, X.; Zhang, X.; Ren, D.; Zhang, X.; Ercolini, D. Structural and Functional Changes in Prokaryotic Communities in Artificial Pit Mud during Chinese Baijiu Production. mSystems 2020, 5, e00829-19. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, R.; Niu, M.; Zheng, J.; Wu, C. Difference of microbial community stressed in artificial pit muds for Luzhou-flavour liquor brewing revealed by multiphase culture-independent technology. J. Appl. Microbiol. 2015, 119, 1345–1356. [Google Scholar] [CrossRef]
- Lu, S.W.; Wei, C.C.; Li, Z.H.; He, X.H.; Tao, Y. Effects of a new caproic acid-producing bacteria on prokaryotic community structure and the acid ester content during the culture of artifcial pit mud. Chin. J. Appl. Environ. Biol. 2020, 26, 144–151. (In Chinese) [Google Scholar]
- Song, J.; Tang, H.; Liang, H.; Luo, L.; Lin, W. Effect of bioaugmentation on biochemical characterisation and microbial communities in Daqu using Bacillus, Saccharomycopsis and Absidia. Int. J. Food Sci. Technol. 2019, 54, 2639–2651. [Google Scholar] [CrossRef]
- Zhang, W.; Si, G.; Du, H.; Li, J.; Zhou, P.; Ye, M. Directional design of a starter to assemble the initial microbial fermentation community of baijiu. Food Res. Int. 2020, 134, 109255. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dong, Y.; Huang, J.; Wang, X.; Zhang, S.; Wu, C.; Jin, Y.; Zhou, R. Alteration of microbial community for improving flavor character of Daqu by inoculation with Bacillus velezensis and Bacillus subtilis. LWT 2019, 111, 1–8. [Google Scholar] [CrossRef]
- He, G.; Huang, J.; Wu, C.; Jin, Y.; Zhou, R. Bioturbation effect of fortified Daqu on microbial community and flavor metabolite in Chinese strong-flavor liquor brewing microecosystem. Food Res. Int. 2020, 129, 108851. [Google Scholar] [CrossRef]
- Zhi, Y.; Wu, Q.; Du, H.; Xu, Y. Biocontrol of geosmin-producing Streptomyces spp. by two Bacillus strains from Chinese liquor. Int. J. Food Microbiol. 2016, 231, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Luo, H.; Yu, D.; Huang, D.; Guo, H.; Zou, Y. Screening and application of biogenic amines degrading strain derived from Luzhou-flavor Daqu. Food Ferment. Ind. 2021, 47, 145–151. (In Chinese) [Google Scholar]
- Cui, K.; Wu, Q.; Xu, Y. Biodegradation of Ethyl Carbamate and Urea with Lysinibacillus sphaericus MT33 in Chinese Liquor Fermentation. J. Agric. Food Chem. 2018, 66, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tian, R.; Wang, K.; Cao, Z.; Yan, P.; Li, F.; Li, X.; Li, S.; He, P. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Zhao, D.; Zhou, R.; Tan, Y.; Wang, T.; Zheng, J. Distribution and function of dominant yeast species in the fermentation of strong-flavor baijiu. World J. Microbiol. Biotechnol. 2021, 37, 26. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, M.; Xu, B.; Liu, L.; Sun, W.; Mu, D.; Wu, X.; Li, X. Microbial communities and flavor formation in the fermentation of Chinese strong-flavor Baijiu produced from old and new Zaopei. Food Res. Int. 2022, 156, 111162. [Google Scholar] [CrossRef]
- Wright, R.J.; Gibson, M.I.; Christie-Oleza, J.A. Understanding microbial community dynamics to improve optimal microbiome selection. Microbiome 2019, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yan, X.; Yang, S.; Chen, F. Screening of Bacillus strains from Luzhou-flavor liquor making for high-yield ethyl hexanoate and low-yield propanol. LWT 2017, 77, 60–66. [Google Scholar] [CrossRef]
- Mao, F.; Huang, J.; Zhou, R.; Qin, H.; Zhang, S.; Cai, X.; Qiu, C. Effects of Daqu properties on the microbial community and their metabolites in fermented grains in Baijiu fermentation system. Can. J. Microbiol. 2023, 69, 170–181. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Zhao, D.; Zheng, J.; Dai, M.; Li, X.; Li, W.; Zhang, C.; Sun, B. Simulated Fermentation of Strong-Flavor Baijiu through Functional Microbial Combination to Realize the Stable Synthesis of Important Flavor Chemicals. Foods 2023, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef]
- Jiao, W.; Xie, F.; Gao, L.; Du, L.; Wei, Y.; Zhou, J.; He, G. Identification of core microbiota in the fermented grains of a Chinese strong-flavor liquor from Sichuan. LWT 2022, 158, 113140. [Google Scholar] [CrossRef]
- Yue, W.; Sun, W.; Rao, R.S.P.; Ye, N.; Yang, Z.; Chen, M. Non-targeted metabolomics reveals distinct chemical compositions among different grades of Bai Mudan white tea. Food Chem. 2019, 277, 289–297. [Google Scholar] [CrossRef]
- Wu, L.; Huang, X.; Liu, S.; Liu, J.; Guo, Y.; Sun, Y.; Lin, J.; Guo, Y.; Wei, S. Understanding the formation mechanism of oolong tea characteristic non-volatile chemical constitutes during manufacturing processes by using integrated widely-targeted metabolome and DIA proteome analysis. Food Chem. 2020, 310, 125941. [Google Scholar] [CrossRef]
- Fan, G.; Liu, P.; Chang, X.; Yin, H.; Cheng, L.; Teng, C.; Gong, Y.; Li, X. Isolation and Identification of a High-Yield Ethyl Caproate-Producing Yeast From Daqu and Optimization of Its Fermentation. Front. Microbiol. 2021, 12, 663744. [Google Scholar] [CrossRef]
- Ran, Y.; Pengxiao, L.; Xu, C.; Jiangqi, X.; Huan, Y.; Guangsen, F.; Chao, T.; Xiuting, L.; Yi, G. Optimization of fermentation conditions for production of ethyl caproate in Baijiu using a selected isolate of Saccharomyces cerevisiae. Emir. J. Food Agric. 2022, 34, 59–69. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Niu, J.; Lin, M.; Zhu, H.; Wang, K.; Li, X.; Sun, B. Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu. Foods 2023, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Luo, L.; Han, Y.; Song, L.; Zhen, P.; Han, D.; Wei, Y.; Zhou, X.; Wen, Z.; Qiu, J.; et al. Microbial Community Affects Daqu Quality and the Production of Ethanol and Flavor Compounds in Baijiu Fermentation. Foods 2023, 12, 2936. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Liu, Y.; Hui, M.; Pan, C. Functional microorganisms in Baijiu Daqu: Research progress and fortification strategy for application. Front. Microbiol. 2023, 14, 1119675. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gong, X.; Zhao, G.; Soe Htet, M.N.; Jia, Z.; Yan, Z.; Liu, L.; Zhai, Q.; Huang, T.; Deng, X.; et al. Liquor Flavour Is Associated with the Physicochemical Property and Microbial Diversity of Fermented Grains in Waxy and Non-waxy Sorghum (Sorghum bicolor) during Fermentation. Front. Microbiol. 2021, 12, 618458. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xiao, X.; Nuccio, E.E.; Yuan, M.; Zhang, N.; Xue, K.; Cohan, F.M.; Zhou, J.; Sun, B. Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environ. Microbiol. 2020, 22, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Huang, Q.; Xiong, Z.; Liao, H.; Lv, Z.; Chen, W.; Luo, X.; Hao, X.; Hug, L.A.; Hug, L.A. Distinct Responses of Rare and Abundant Microbial Taxa to In Situ Chemical Stabilization of Cadmium-Contaminated Soil. mSystems 2021, 6, e01040-21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xing, J.; Gu, S.; Wu, Y.; Li, X.; Ma, J.; Mao, J. Distinct succession of abundant and rare fungi in fermented grains during Chinese strong-flavor liquor fermentation. LWT 2022, 163, 113502. [Google Scholar] [CrossRef]
- Shang, C.; Li, Y.; Zhang, J.; Gan, S. Analysis of Bacterial Diversity in Different Types of Daqu and Fermented Grains from Danquan Distillery. Front. Microbiol. 2022, 13, 883122. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Dong, S.; Zhou, Z.; Zhang, Z.; Huang, R.; Han, S.; Hou, J.; Pan, C. Dynamic changes and correlations of microbial communities, physicochemical properties, and volatile metabolites during Daqu fermentation of Taorong-type Baijiu. LWT 2023, 173, 114290. [Google Scholar] [CrossRef]
- Kamli, M.R.; Alzahrani, N.A.Y.; Hajrah, N.H.; Sabir, J.S.M.; Malik, A. Genome-Driven Discovery of Enzymes with Industrial Implications from the Genus Aneurinibacillus. Microorganisms 2021, 9, 499. [Google Scholar] [CrossRef]
- Liu, W.; Chai, L.; Wang, H.; Lu, Z.; Zhang, X.; Xiao, C.; Wang, S.; Shen, C.; Shi, J.; Xu, Z. Bacteria and filamentous fungi running a relay race in Daqu fermentation enable macromolecular degradation and flavor substance formation. Int. J. Food Microbiol. 2023, 390, 110118. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Xue, Y.A.; Wang, Y.; Wang, W.; Shu, N.; Zhao, H.; Tang, F.; Yang, X.; Guo, Z.; Shan, C. The Fungal Communities and Flavor Profiles in Different Types of High-Temperature Daqu as Revealed by High-Throughput Sequencing and Electronic Senses. Front. Microbiol. 2021, 12, 784651. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhu, M.; Du, Y.; Wu, Z.; Gomi, K.; Zhang, W. Metaproteomics reveals protein composition of multiple saccharifying enzymes in nongxiangxing daqu and jiangxiangxing daqu under different thermophilic temperatures. Int. J. Food Sci. Technol. 2022, 57, 5102–5113. [Google Scholar] [CrossRef]
- Li, X.; Tan, G.; Chen, P.; Cai, K.; Dong, W.; Peng, N.; Zhao, S. Uncovering acid resistance genes in lactic acid bacteria and impact of non-viable bacteria on bacterial community during Chinese strong-flavor baijiu fermentation. Food Res. Int. 2023, 167, 112741. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, K.; Chen, M.; Fan, J.; Han, S.; Hou, J.; Chi, L.; Liu, Y.; Wei, T. Profiling the composition and metabolic activities of microbial community in fermented grain for the Chinese strong-flavor Baijiu production by using the metatranscriptome, high-throughput 16S rRNA and ITS gene sequencings. Food Res. Int. 2020, 138, 109765. [Google Scholar] [CrossRef] [PubMed]
- Iacono, R.; De Lise, F.; Moracci, M.; Cobucci-Ponzano, B.; Strazzulli, A. Glycoside hydrolases from (hyper)thermophilic archaea: Structure, function, and applications. Essays Biochem. 2023, 67, 731–751. [Google Scholar] [PubMed]
- Zabotina, O.A.; Zhang, N.; Weerts, R. Polysaccharide Biosynthesis: Glycosyltransferases and Their Complexes. Front. Plant Sci. 2021, 12, 625307. [Google Scholar] [CrossRef]
- He, M.; Jin, Y.; Zhou, R.; Zhao, D.; Zheng, J.; Wu, C. Dynamic succession of microbial community in Nongxiangxing daqu and microbial roles involved in flavor formation. Food Res. Int. 2022, 159, 111559. [Google Scholar] [CrossRef]
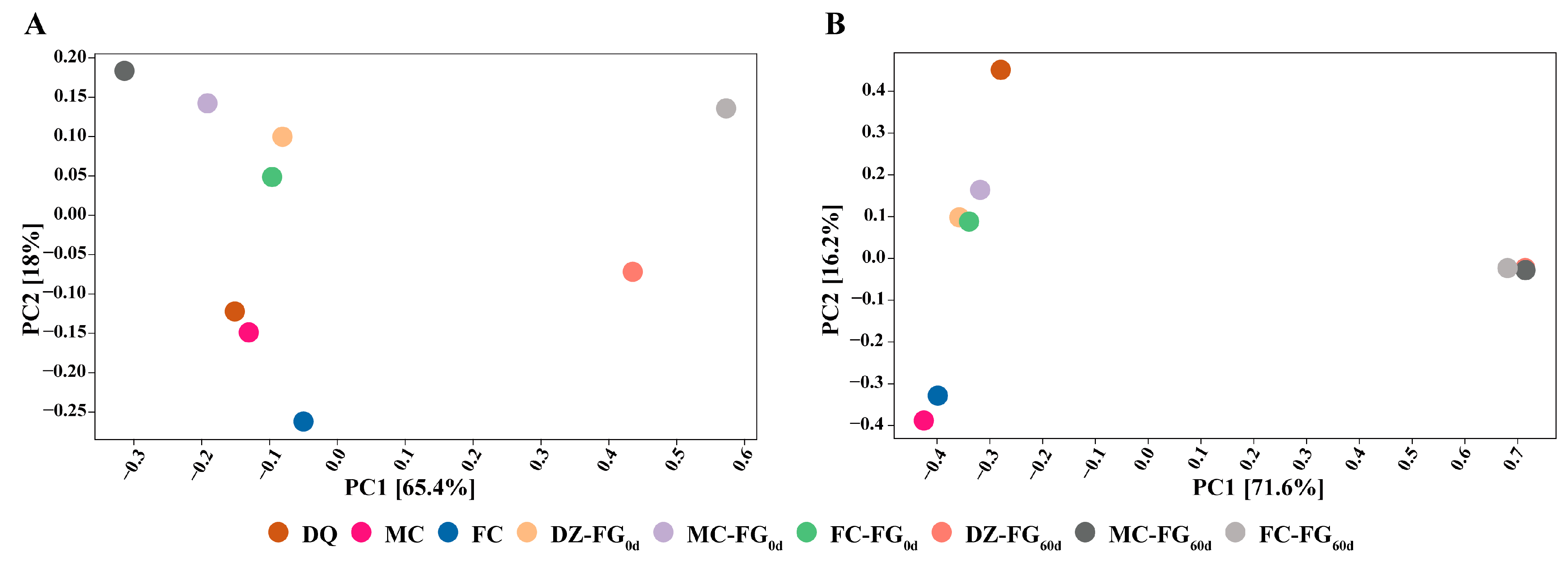

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | The Date of Samples | The Ratios of Daqu or PM Cultures (w/w) | ||
|---|---|---|---|---|
| Daqu (DQ) | MC | FC | ||
| DZ-FG0d | The 0 days of DZ-FG | 15% | - | - |
| MC-FG0d | The 0 days of MC-FG | 15% | 1% | - |
| FC-FG0d | The 0 days of FC-FG | 15% | - | 1% |
| DZ-FG30d | The 30th day of DZ-FG | 15% | - | - |
| MC-FG30d | The 30th day of MC-FG | 15% | 1% | - |
| FC-FG30d | The 30th day of FC-FG | 15% | - | 1% |
| DZ-FG60d | The 60th day of DZ-FG | 15% | - | - |
| MC-FG60d | The 60th day of MC-FG | 15% | 1% | - |
| FC-FG60d | The 60th day of FC-FG | 15% | - | 1% |
| Sample | Moisture (%) | Starch (g/100 g) | Reducing Sugar (g/100 g) | Acidity (mmol/10 g) | Alcohol (%) |
|---|---|---|---|---|---|
| DZ-FG0d | 57.96 ± 0.26 | 23.45 ± 0.24 a | 1.55 ± 0.06 a | 0.85 ± 0.00 b | ND |
| MC-FG0d | 57.53 ± 0.32 | 22.66 ± 0.22 b | 1.33 ± 0.03 b | 0.92 ± 0.04 ab | ND |
| FC-FG0d | 57.33 ± 0.00 | 23.90 ± 0.25 a | 1.39 ± 0.09 b | 0.95 ± 0.03 a | ND |
| DZ-FG30d | 64.61 ± 0.14 | 13.97 ± 0.45 b | 1.15 ± 0.02 c | 1.00 ± 0.01 b | 8.43 ± 0.45 a |
| MC-FG30d | 63.55 ± 0.23 | 15.27 ± 0.05 a | 1.37 ± 0.06 a | 1.09 ± 0.02 a | 7.20 ± 0.50 b |
| FC-FG30d | 64.22 ± 0.57 | 15.86 ± 0.16 a | 1.25 ± 0.00 b | 1.00 ± 0.01 b | 7.70 ± 0.50 ab |
| DZ-FG60d | 65.91 ± 0.53 | 13.16 ± 0.50 a | 1.18 ± 0.03 b | 1.88 ± 0.01 b | 7.00 ± 0.20 |
| MC-FG60d | 64.52 ± 0.90 | 14.33 ± 0.26 a | 0.84 ± 0.00 c | 2.11 ± 0.03 a | 6.47 ± 0.15 |
| FC-FG60d | 63.92 ± 0.34 | 11.60 ± 1.14 b | 1.60 ± 0.01 a | 1.83 ± 0.01 b | 6.70 ± 0.50 |
| Sample | Bacteria | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|
| Chao1 | Observed Species | Shannon | Simpson | Chao1 | Observed Species | Shannon | Simpson | |
| DQ | 234.06 | 227.60 | 4.08 | 0.84 | 69.49 | 68.50 | 1.85 | 0.60 |
| MC | 216.87 | 215.00 | 3.58 | 0.77 | 95.76 | 94.40 | 2.44 | 0.66 |
| FC | 141.16 | 141.00 | 3.59 | 0.87 | 44.80 | 44.30 | 2.38 | 0.75 |
| DZ-FG0d | 131.23 | 130.50 | 4.60 | 0.92 | 63.17 | 62.10 | 2.24 | 0.64 |
| MC-FG0d | 143.97 | 142.80 | 4.78 | 0.93 | 65.51 | 63.20 | 1.84 | 0.50 |
| FC-FG0d | 181.07 | 161.30 | 3.41 | 0.74 | 71.67 | 69.60 | 2.44 | 0.66 |
| DZ-FG60d | 52.39 | 46.20 | 1.44 | 0.55 | 49.14 | 45.90 | 2.50 | 0.74 |
| MC-FG60d | 40.24 | 39.80 | 1.84 | 0.62 | 54.23 | 52.60 | 1.81 | 0.46 |
| FC-FG60d | 47.07 | 45.70 | 1.57 | 0.51 | 37.03 | 37.00 | 2.13 | 0.68 |
| Classification | AAT | CAT | CRAT | ART | CRT | MT | Abundant Taxa | Rare Taxa | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | Daqu and PM cultures | ASV proportion | 0.84% | 0.84% | 9.24% | 18.49% | 68.07% | 2.52% | 10.92% | 86.55% |
| Relative abundance | 41.31% | 1.04% | 52.04% | 0.05% | 5.11% | 0.44% | 94.39% | 5.17% | ||
| FG0d | ASV proportion | 7.29% | 3.13% | 1.04% | 23.96% | 43.75% | 20.83% | 11.46% | 67.71% | |
| Relative abundance | 91.03% | 2.25% | 0.96% | 0.03% | 1.35% | 4.38% | 94.24% | 1.38% | ||
| FG60d | ASV proportion | 2.22% | 0.00% | 4.44% | 60.00% | 31.11% | 2.22% | 6.67% | 91.11% | |
| Relative abundance | 97.88% | 0.00% | 1.43% | 0.06% | 0.30% | 0.34% | 99.31% | 0.36% | ||
| Fungi | Daqu and PM cultures | ASV proportion | 3.70% | 5.56% | 5.56% | 33.33% | 44.44% | 7.41% | 14.81% | 77.78% |
| Relative abundance | 76.29% | 16.78% | 5.30% | 0.04% | 1.10% | 0.49% | 98.37% | 1.14% | ||
| FG0d | ASV proportion | 8.70% | 6.52% | 0.00% | 52.17% | 23.91% | 8.70% | 15.22% | 76.09% | |
| Relative abundance | 96.92% | 2.27% | 0.00% | 0.05% | 0.27% | 0.49% | 99.19% | 0.32% | ||
| FG60d | ASV proportion | 2.63% | 18.42% | 7.89% | 50.00% | 21.05% | 0.00% | 28.95% | 71.05% | |
| Relative abundance | 37.83% | 56.52% | 5.37% | 0.04% | 0.24% | 0.00% | 99.72% | 0.28% |
| Top Ten Genera Based on Relative Abundance | Top Ten Species Based on Relative Abundance | ||||||
|---|---|---|---|---|---|---|---|
| Genus | DZ-FG60d | MC-FG60d | FC-FG60d | Species | DZ-FG60d | MC-FG60d | FC-FG60d |
| Lactobacillus | 97.85% | 33.93% | 97.74% | Lactobacillus acetotolerans | 96.20% | 33.72% | 96.08% |
| Acetilactobacillus | 1.17% | 65.54% | 1.06% | Acetilactobacillus jinshanensis | 1.18% | 65.54% | 1.06% |
| Pichia | 0.24% | 0.05% | 0.32% | Lactobacillus helveticus | 0.74% | 0.04% | 0.74% |
| Saccharomyces | 0.12% | 0.01% | 0.22% | Lactobacillus amylolyticus | 0.42% | 0.02% | 0.41% |
| Lentilactobacillus | 0.13% | 0.02% | 0.13% | Lactobacillus crispatus | 0.32% | 0.12% | 0.31% |
| Acetobacter | 0.03% | 0.17% | 0.03% | Pichia kudriavzevii | 0.24% | 0.05% | 0.32% |
| Ligilactobacillus | 0.11% | 0.02% | 0.11% | Saccharomyces cerevisiae | 0.12% | 0.01% | 0.22% |
| Kroppenstedtia | 0.08% | 0.04% | 0.08% | Ligilactobacillus acidipiscis | 0.11% | 0.02% | 0.11% |
| Loigolactobacillus | 0.05% | 0.03% | 0.05% | Lentilactobacillus buchneri | 0.11% | 0.01% | 0.11% |
| Sphingomonas | 0.04% | 0.02% | 0.05% | Lactobacillus amylovorus | 0.09% | 0.01% | 0.12% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Y.; Huang, J.; Tang, Q.; Zhang, S.; Qin, H.; Dong, Y.; Wang, X.; Qiu, C.; Huang, M.; Zhang, Z.; et al. Characterizing the Contribution of Functional Microbiota Cultures in Pit Mud to the Metabolite Profiles of Fermented Grains. Foods 2024, 13, 1597. https://doi.org/10.3390/foods13111597
Wan Y, Huang J, Tang Q, Zhang S, Qin H, Dong Y, Wang X, Qiu C, Huang M, Zhang Z, et al. Characterizing the Contribution of Functional Microbiota Cultures in Pit Mud to the Metabolite Profiles of Fermented Grains. Foods. 2024; 13(11):1597. https://doi.org/10.3390/foods13111597
Chicago/Turabian StyleWan, Yingdong, Jun Huang, Qiuxiang Tang, Suyi Zhang, Hui Qin, Yi Dong, Xiaojun Wang, Chuanfeng Qiu, Mengyang Huang, Zhu Zhang, and et al. 2024. "Characterizing the Contribution of Functional Microbiota Cultures in Pit Mud to the Metabolite Profiles of Fermented Grains" Foods 13, no. 11: 1597. https://doi.org/10.3390/foods13111597
APA StyleWan, Y., Huang, J., Tang, Q., Zhang, S., Qin, H., Dong, Y., Wang, X., Qiu, C., Huang, M., Zhang, Z., Zhang, Y., & Zhou, R. (2024). Characterizing the Contribution of Functional Microbiota Cultures in Pit Mud to the Metabolite Profiles of Fermented Grains. Foods, 13(11), 1597. https://doi.org/10.3390/foods13111597