
Improving Catalytic Efficiency of L-Arabinose Isomerase from Lactobacillus plantarum CY6 towards D-Galactose by Molecular Modification

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Materials, and Plasmid Construction
2.2. Structural Modeling and In Silico Mutagenesis of LpAI
2.3. Site-Directed Mutagenesis
2.4. Protein Expression and Purification
2.5. Determination of Enzyme Activity and Kinetic Parameter
2.6. Effect of Temperature, pH, and Divalent Metal Ions
2.7. D-Tagatose Biosynthesis of Whole Cell
2.8. Molecular Docking Analysis
2.9. Statistical Analysis
3. Results
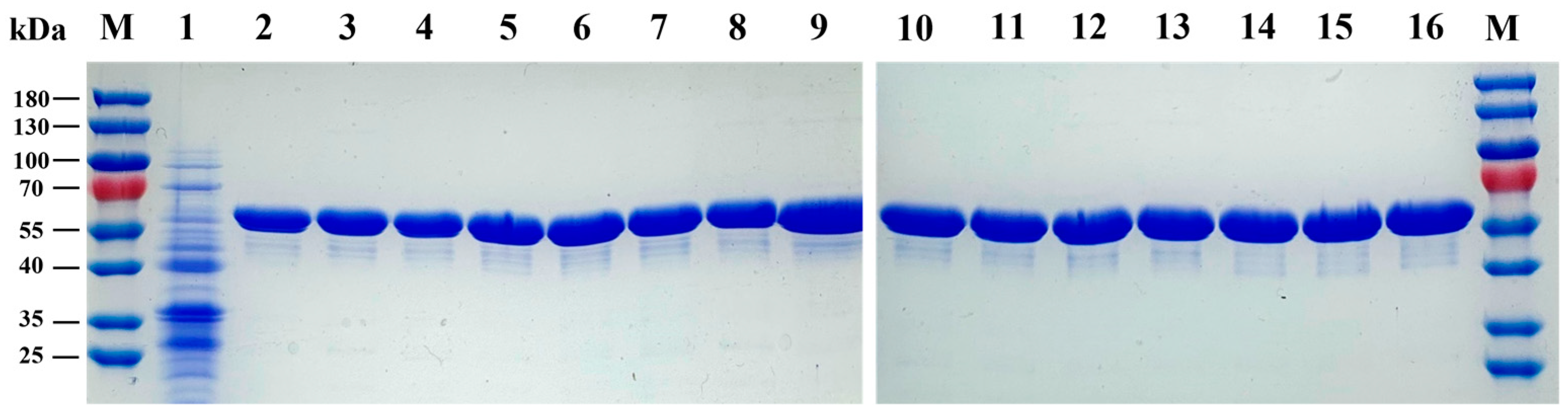
3.1. Expression and Purification of LpAI
3.2. Biochemical Characterization of LpAI
3.2.1. Effect of Metal Ion
3.2.2. Effect of Temperature and pH
3.3. Rational Design to Develop a Highly D-Galactose Isomerizing LpAI
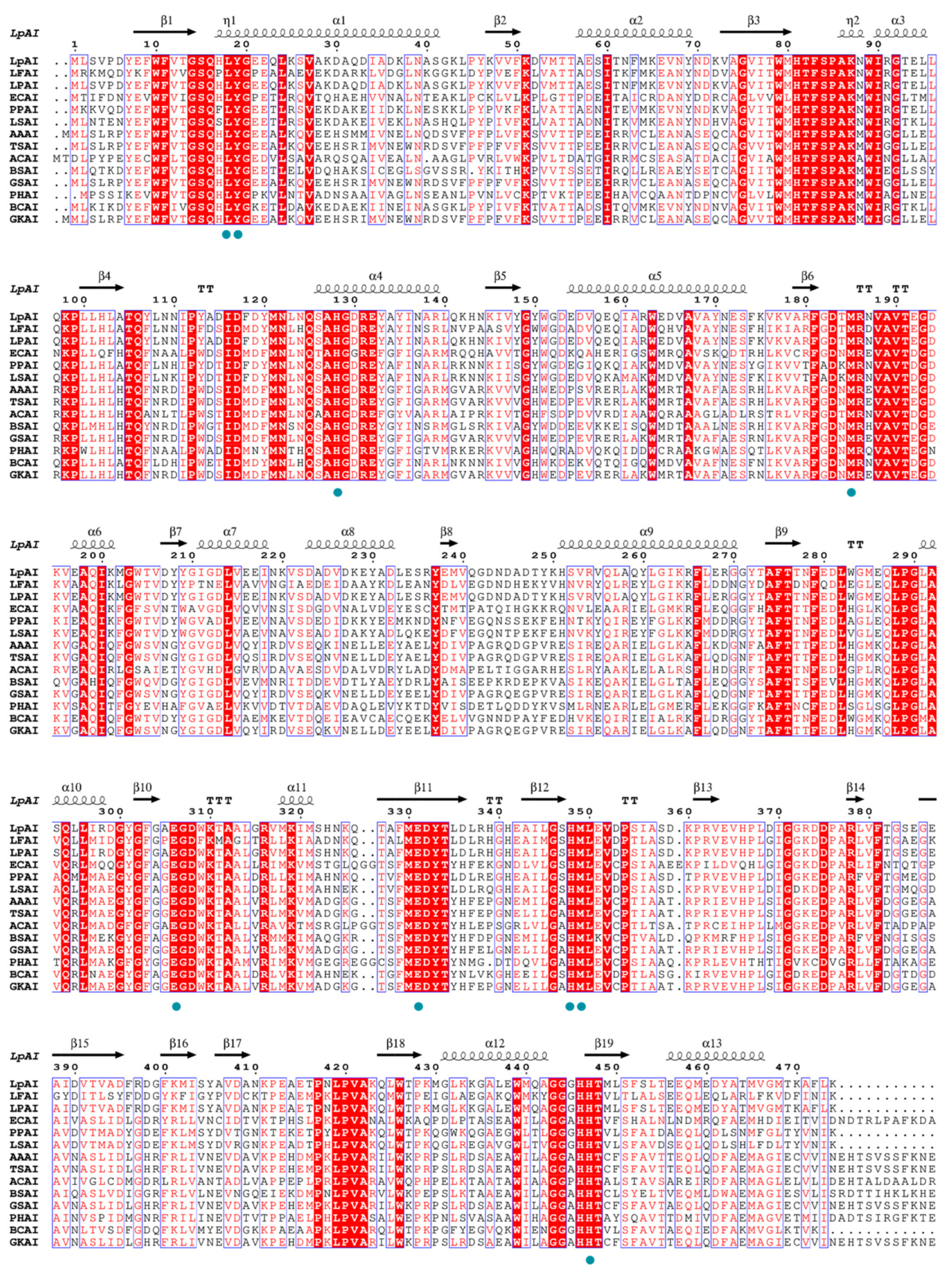
3.3.1. Multiple Sequence Alignment, Homology Modeling, and Active Site Analysis
3.3.2. Molecular Modification of LpAI
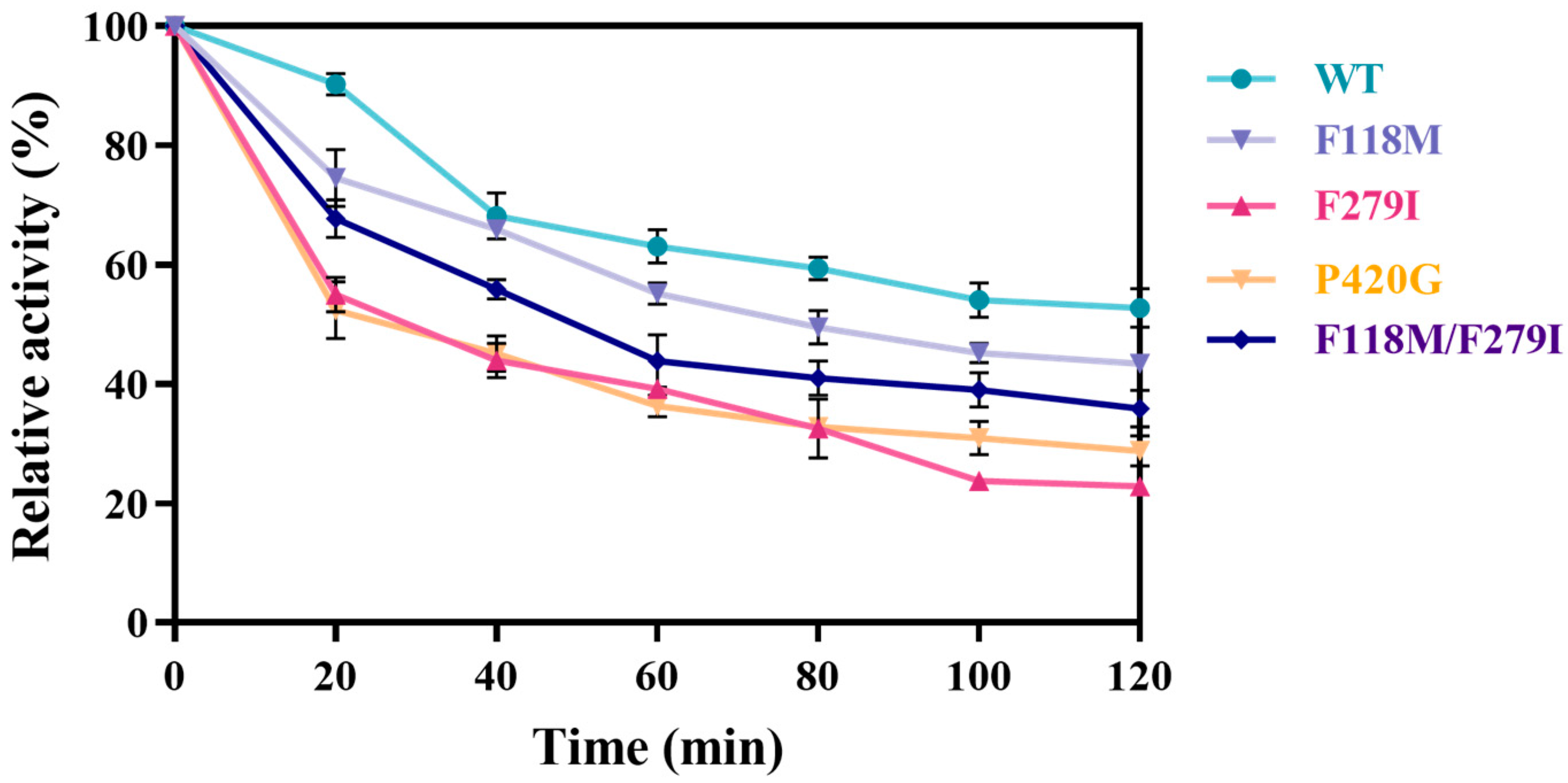
3.3.3. Iterative Mutation Characterization and Kinetic Parameters
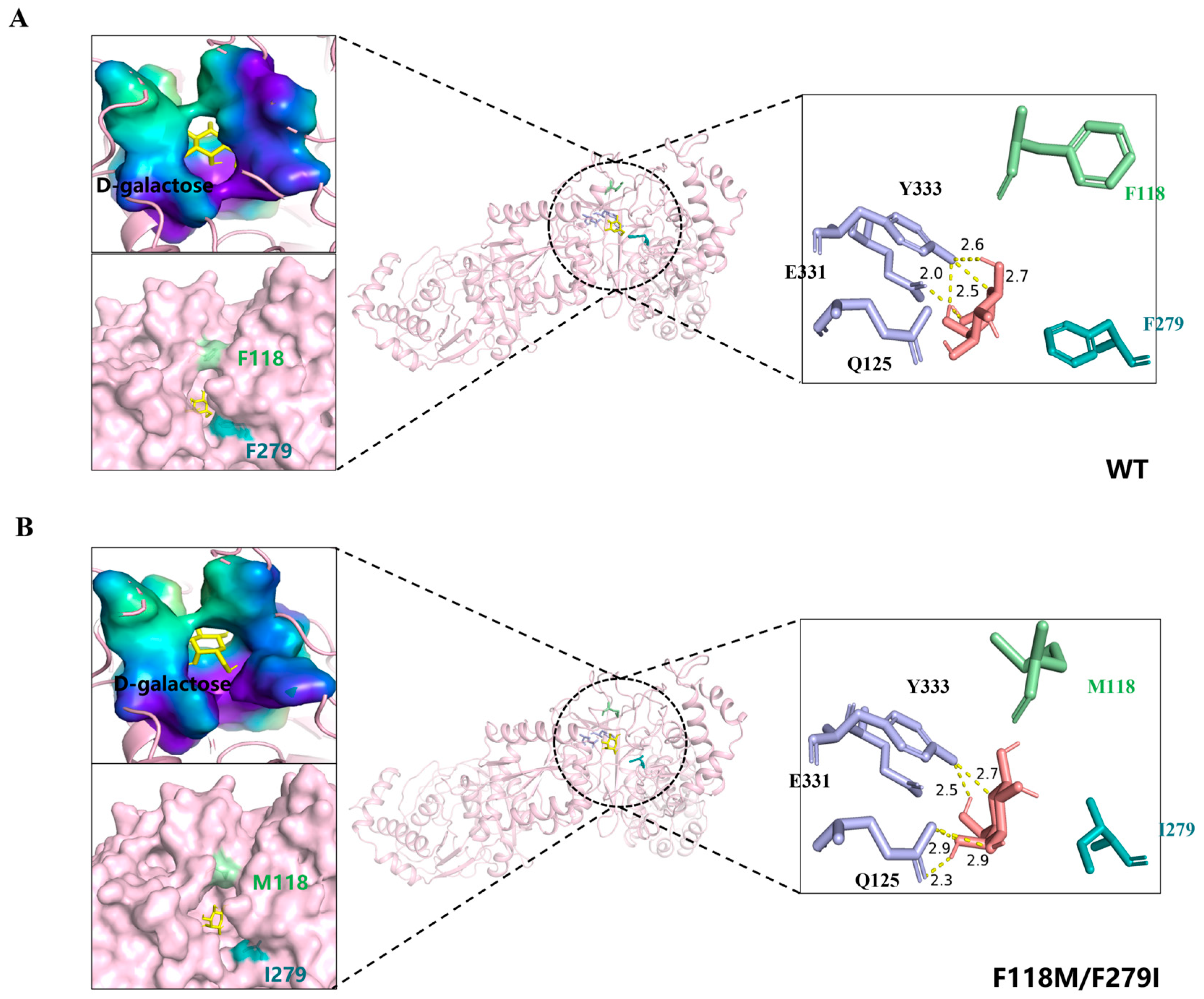
3.4. Molecular Docking and Whole Cell Biocatalytic Synthesis of D-Tagatose
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ravikumar, Y.; Ponpandian, L.N.; Zhang, G.; Yun, J.; Qi, X. Harnessing L-Arabinose Isomerase for Biological Production of D-Tagatose: Recent Advances and Its Applications. Trends Food Sci. Technol. 2021, 107, 16–30. [Google Scholar] [CrossRef]
- Levin, G.; Zehner, L.; Saunders, J.; Beadle, J. Sugar Substitutes: Their Energy Values, Bulk Characteristics, and Potential Health Benefits. Am. J. Clin. Nutr. 1995, 62, 1161S–1168S. [Google Scholar] [CrossRef]
- Nagamine, Y.; Hasibul, K.; Ogawa, T.; Tada, A.; Kamitori, K.; Hossain, A.; Yamaguchi, F.; Tokuda, M.; Kuwahara, T.; Miyake, M. D-Tagatose Effectively Reduces the Number of Streptococcus Mutans and Oral Bacteria in Healthy Adult Subjects: A Chewing Gum Pilot Study and Randomized Clinical Trial. Acta Medica Okayama 2020, 74, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Levin, G.V. Tagatose, the New GRAS Sweetener and Health Product. J. Med. Food. 2002, 5, 23–36. [Google Scholar] [CrossRef]
- Oh, D.K. Tagatose: Properties, Applications, and Biotechnological Processes. Appl. Microbiol. Biotechnol. 2007, 76, 1–8. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, W.; Zhang, T.; Jiang, B.; Mu, W. L-Arabinose Isomerases: Characteristics, Modification, and Application. Trends Food Sci. Technol. 2018, 78, 25–33. [Google Scholar] [CrossRef]
- Beadle, J.; Saunders, J.; Wajda, T. Process of Manufacturing Tagatose. U.S. Patent 5078796A, 7 January 1992. Available online: https://www.google.ch/patents/US5078796 (accessed on 28 October 2023).
- Oh, D.K.; Kim, H.J.; Ryu, S.A. Development of An Immobilization Method of L-Arabinose Isomerase for Industrial Production of Tagatose. Biotechnol. Lett. 2001, 23, 1859–1862. [Google Scholar] [CrossRef]
- Jayamuthunagai, J.; Gautam, P.; Srisowmeya, G.; Chakravarthy, M. Biocatalytic Production of D-Tagatose: A Potential Rare Sugar with Versatile Applications. Crit. Rev. Food Sci. Nutr. 2017, 57, 3430–3437. [Google Scholar] [CrossRef]
- Rhimi, M.; Bejar, S. Cloning, Purification and Biochemical Characterization of Metallic-Ions Independent and Thermoactive L-Arabinose Isomerase from the Bacillus stearothermophilus US100 Strain. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2006, 1760, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Lee, Y.J.; Cao, T.P.; Shin, S.-M.; Park, M.K.; Lee, H.S.; Luccio, E.; Kim, S.B.; Lee, S.J.; Lee, S.J. Structure of the Thermophilic L-Arabinose Isomerase from Geobacillus kaustophilus Reveals Metal-Mediated Intersubunit Interactions for Activity and Thermostability. Arch. Biochem. Biophys. 2016, 596, 51–62. [Google Scholar] [CrossRef]
- Yuan, J.; Ravikumar, Y.; Zhang, G.; Yun, J.; Zhang, Y.; Zabed, H.M.; Qi, X. L-Arabinose Isomerase from Lactobacillus parabuchneri and Its Whole Cell Biocatalytic Application in D-Tagatose Biosynthesis from D-Galactose. Food Biosci. 2021, 41, 101034. [Google Scholar] [CrossRef]
- Fan, C.; Liu, K.; Zhang, T.; Zhou, L.; Xue, D.; Jiang, B.; Mu, W. Biochemical Characterization of a Thermostable L-Arabinose Isomerase from a Thermoacidophilic Bacterium, Alicyclobacillus hesperidum URH17-3-68. J. Mol. Catal. B 2014, 102, 120–126. [Google Scholar] [CrossRef]
- Oh, H.J.; Kim, H.J.; Oh, D.K. Increase in D-Tagatose Production Rate by Site-Directed Mutagenesis of L-Arabinose Isomerase from Geobacillus thermodenitrificans. Biotechnol. Lett. 2006, 28, 145–149. [Google Scholar] [CrossRef]
- Cheng, L.; Mu, W.; Zhang, T.; Jiang, B. An L-Arabinose Isomerase from Acidothermus cellulolytics ATCC 43068: Cloning, Expression, Purification, and Characterization. Appl. Microbiol. Biotechnol. 2010, 86, 1089–1097. [Google Scholar] [CrossRef]
- Hong, Y.H.; Lee, D.W.; Lee, S.J.; Choe, E.A.; Kim, S.B.; Lee, Y.H.; Cheigh, C.I.; Pyun, Y.R. Production of D-Tagatose at High Temperatures Using Immobilized Escherichia Coli Cells Expressing L-Arabinose Isomerase from Thermotoga neapolitana. Biotechnol. Lett. 2007, 29, 569–574. [Google Scholar] [CrossRef]
- Xu, Z.; Qing, Y.; Li, S.; Feng, X.; Xu, H.; Ouyang, P. A Novel L-Arabinose Isomerase from Lactobacillus fermentum CGMCC2921 for D-Tagatose Production: Gene Cloning, Purification and Characterization. J. Mol. Catal. B 2011, 70, 1–7. [Google Scholar] [CrossRef]
- Laksmi, F.A.; Arai, S.; Tsurumaru, H.; Nakamura, Y.; Saksono, B.; Tokunaga, M.; Ishibashi, M. Improved Substrate Specificity for D-Galactose of L-Arabinose Isomerase for Industrial Application. BBA-Proteins Proteom. 2018, 1866, 1084–1091. [Google Scholar] [CrossRef]
- Xu, Z.; Li, S.; Feng, X.; Zhan, Y.; Xu, H. Function of Aspartic Acid Residues in Optimum pH Control of L-Arabinose Isomerase from Lactobacillus fermentum. Appl. Microbiol. Biotechnol. 2014, 98, 3987–3996. [Google Scholar] [CrossRef]
- Zhang, G.; Zabed, H.M.; Yun, J.; Yuan, J.; Zhang, Y.; Wang, Y.; Qi, X. Two-Stage Biosynthesis of D-Tagatose from Milk Whey Powder by an Engineered Escherichia Coli Strain Expressing L-Arabinose Isomerase from Lactobacillus plantarum. Bioresour. Technol. 2020, 305, 123010. [Google Scholar] [CrossRef]
- Pucci, F.; Rooman, M. Improved Insights into Protein Thermal Stability: From the Molecular to the Structurome Scale. Philos. Trans. R. Soc. A 2016, 374, 20160141. [Google Scholar] [CrossRef]
- Zheng, Z.; Mei, W.; Xia, M.; He, Q.; Ouyang, J. Rational Design of Bacillus coagulans NL01 L-Arabinose Isomerase and Use of Its F279I Variant in d-Tagatose Production. J. Agric. Food Chem. 2017, 65, 4715–4721. [Google Scholar] [CrossRef]
- Jayaraman, A.B.; Kandasamy, T.; Venkataraman, D.; Meenakshisundaram, S. Rational Design of Shewanella Sp. l-Arabinose Isomerase for D-Galactose Isomerase Activity under Mesophilic Conditions. Enzyme Microb. Technol. 2021, 147, 109796. [Google Scholar] [CrossRef]
- Kim, B.J.; Hong, S.H.; Shin, K.C.; Jo, Y.S.; Oh, D.K. Characterization of a F280N Variant of L-Arabinose Isomerase from Geobacillus thermodenitrificans Identified as a D-Galactose Isomerase. Appl. Microbiol. Biotechnol. 2014, 98, 9271–9281. [Google Scholar] [CrossRef]
- Li, L.; Yu, W.; Jia, P.; Li, C.; Jing, X.; Zhang, L.; Lv, H. Method for Purifying Tagatose by Using Sequential Simulated Moving Bed Chromatography (SSMB). C.N. Patent CN103992362A, 12 June 2014. [Google Scholar]
- Ma, Y.; Shi, T.; Li, Y. Method for Preparing High-Purity Crystallized D-Tagatose. C.N. Patent CN112592378A, 1 June 2021. [Google Scholar]
- Liang, M.; Chen, M.; Liu, X.; Zhai, Y.; Liu, X.; Zhang, H.; Xiao, M.; Wang, P. Bioconversion of D-galactose to D-tagatose: Continuous packed bed reaction with an immobilized thermostable L-Arabinose isomerase and efficient purification by selective microbial degradation. Appl. Microbiol. Biotechnol. 2012, 93, 1469–1474. [Google Scholar] [CrossRef]
- Zheng, Z.; Xie, J.; Liu, P.; Li, X.; Ouyang, J. Elegant and Efficient Biotransformation for Dual Production of d-Tagatose and Bioethanol from Cheese Whey Powder. J. Agric. Food Chem. 2019, 67, 829–835. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km (mM) | Vmax (U mg−1) | Kcat (min−1) | Kcat/Km (mM−1 min−1) |
|---|---|---|---|---|
| WT | 1338.01 | 7.27 | 227.19 | 0.17 |
| F118M | 1229.88 | 9.03 | 334.33 | 0.27 |
| F279I | 1271.62 | 16.46 | 576.59 | 0.45 |
| P420G | 1305.30 | 8.31 | 230.75 | 0.18 |
| F118M/F279I | 1013.05 | 20.88 | 535.38 | 0.53 |
| Enzyme | Specific Activity (U mg−1) | Ratio a (%) | |
|---|---|---|---|
| D-Galactose | L-Arabinose | ||
| WT | 2.37 | 3.79 | 62.53 |
| F118M | 3.25 | 3.53 | 92.07 |
| F279I | 5.00 | 3.35 | 149.25 |
| P420G | 2.94 | 3.73 | 78.82 |
| F118M/F279I | 7.35 | 3.46 | 212.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Chen, Z.; Ravikumar, Y.; Zhang, G.; Tang, X.; Zhang, Y.; Zhao, M.; Sun, W.; Qi, X. Improving Catalytic Efficiency of L-Arabinose Isomerase from Lactobacillus plantarum CY6 towards D-Galactose by Molecular Modification. Foods 2024, 13, 1727. https://doi.org/10.3390/foods13111727
Lu C, Chen Z, Ravikumar Y, Zhang G, Tang X, Zhang Y, Zhao M, Sun W, Qi X. Improving Catalytic Efficiency of L-Arabinose Isomerase from Lactobacillus plantarum CY6 towards D-Galactose by Molecular Modification. Foods. 2024; 13(11):1727. https://doi.org/10.3390/foods13111727
Chicago/Turabian StyleLu, Chengyu, Ziwei Chen, Yuvaraj Ravikumar, Guoyan Zhang, Xinrui Tang, Yufei Zhang, Mei Zhao, Wenjing Sun, and Xianghui Qi. 2024. "Improving Catalytic Efficiency of L-Arabinose Isomerase from Lactobacillus plantarum CY6 towards D-Galactose by Molecular Modification" Foods 13, no. 11: 1727. https://doi.org/10.3390/foods13111727