
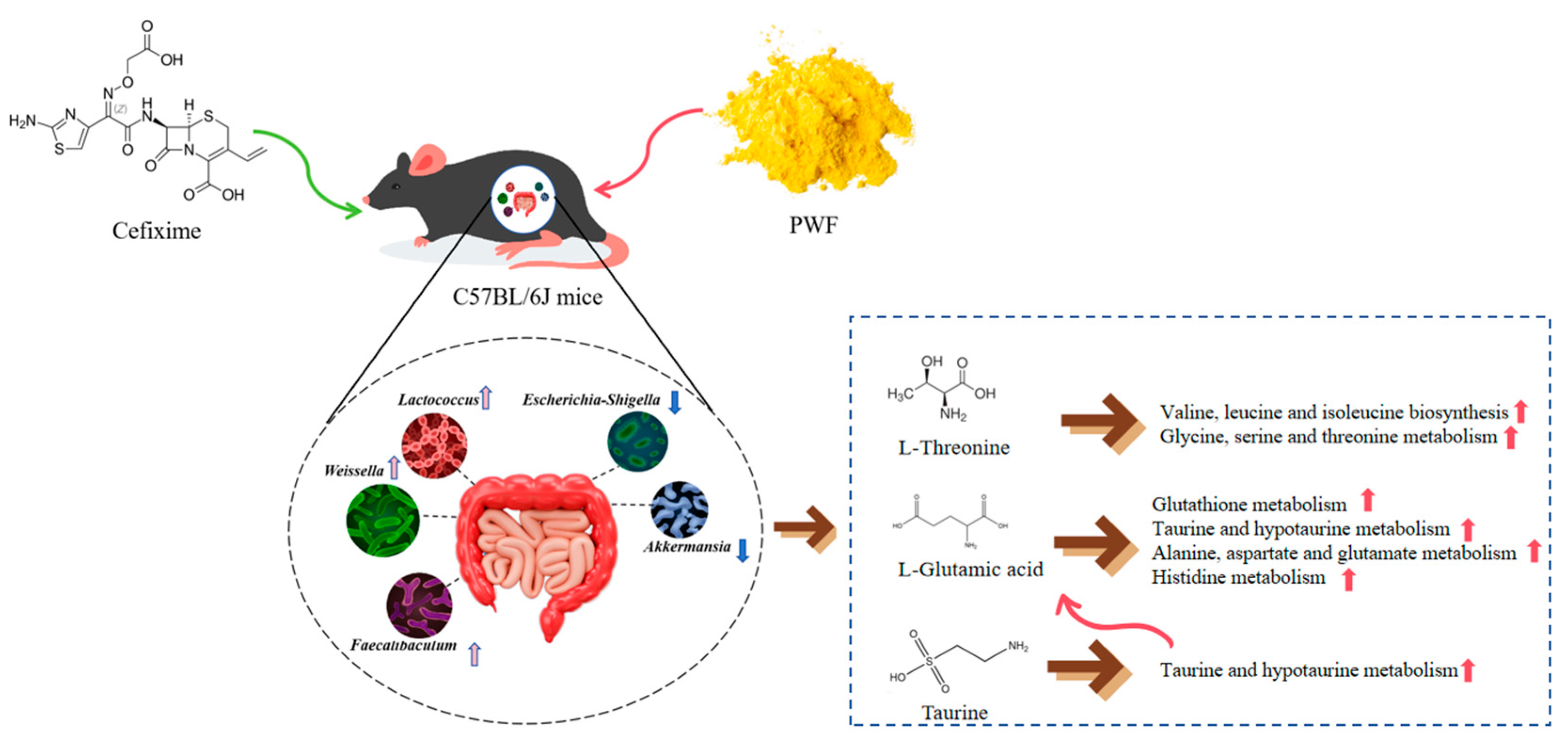
Investigating the Impact of Pineapple–Whey Protein Fermentation Products on Cefixime-Induced Intestinal Flora Dysbiosis in Mice Using 16S Sequencing and Untargeted Metabolomics Techniques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Collection
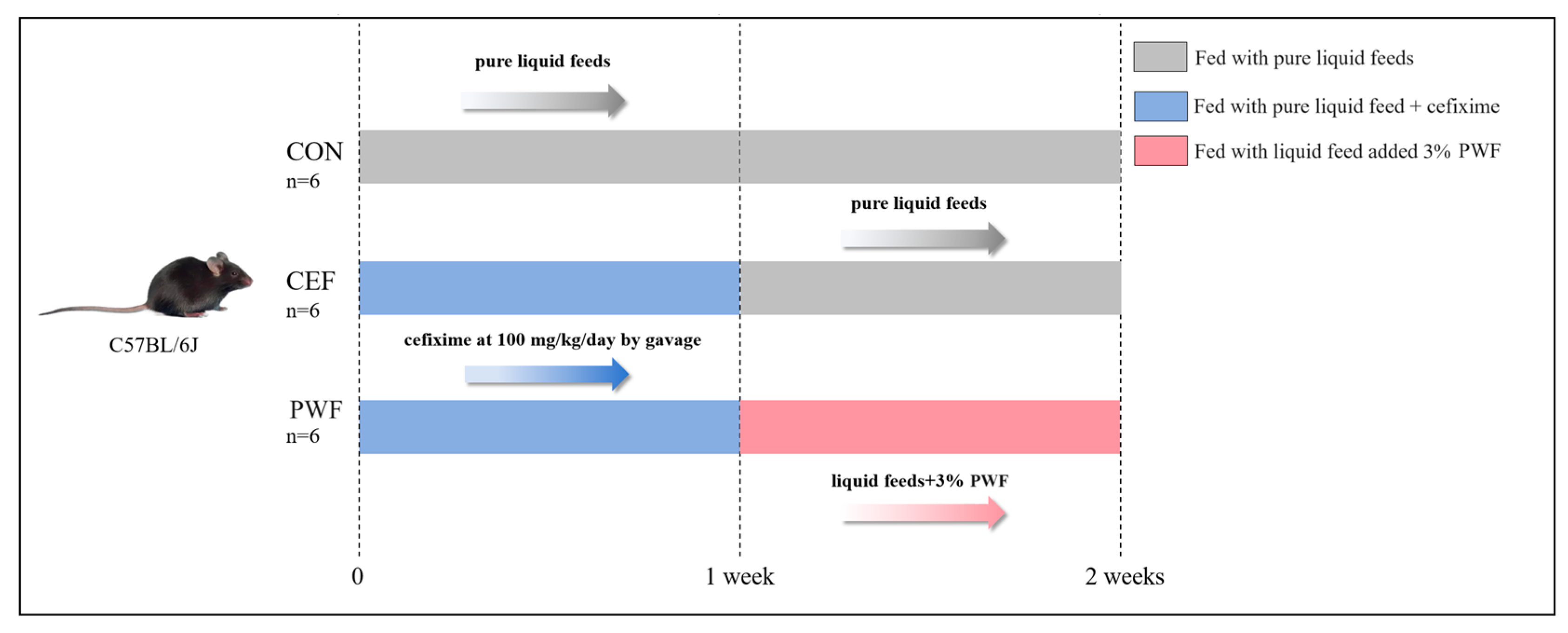
2.2. Animal Treatments
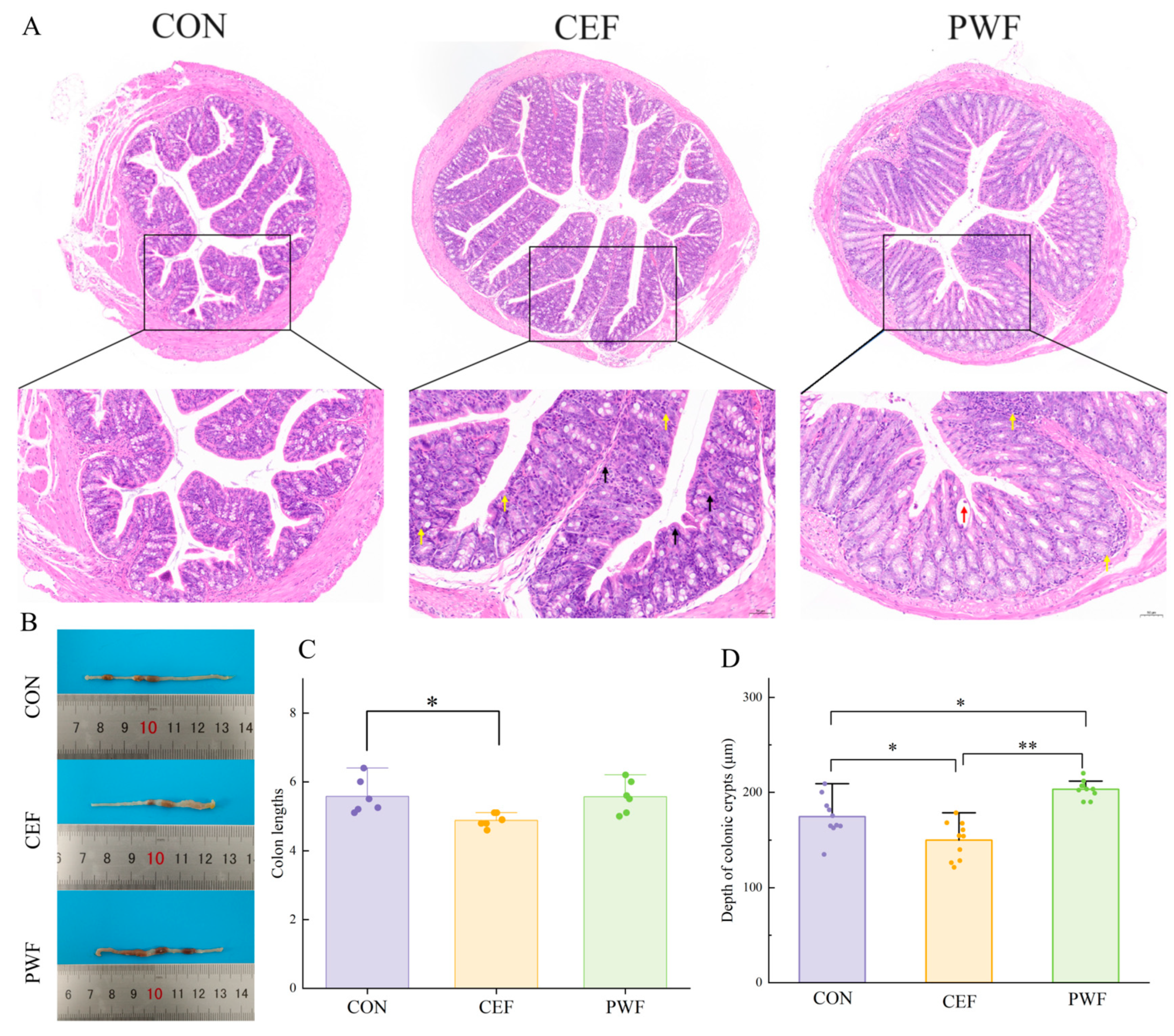
2.3. Histomorphological Analysis of Colons
2.4. Analysis of Gut Microflora
2.5. Analysis of the Metabolome
2.6. Statistical Analysis
3. Results
3.1. Subsection
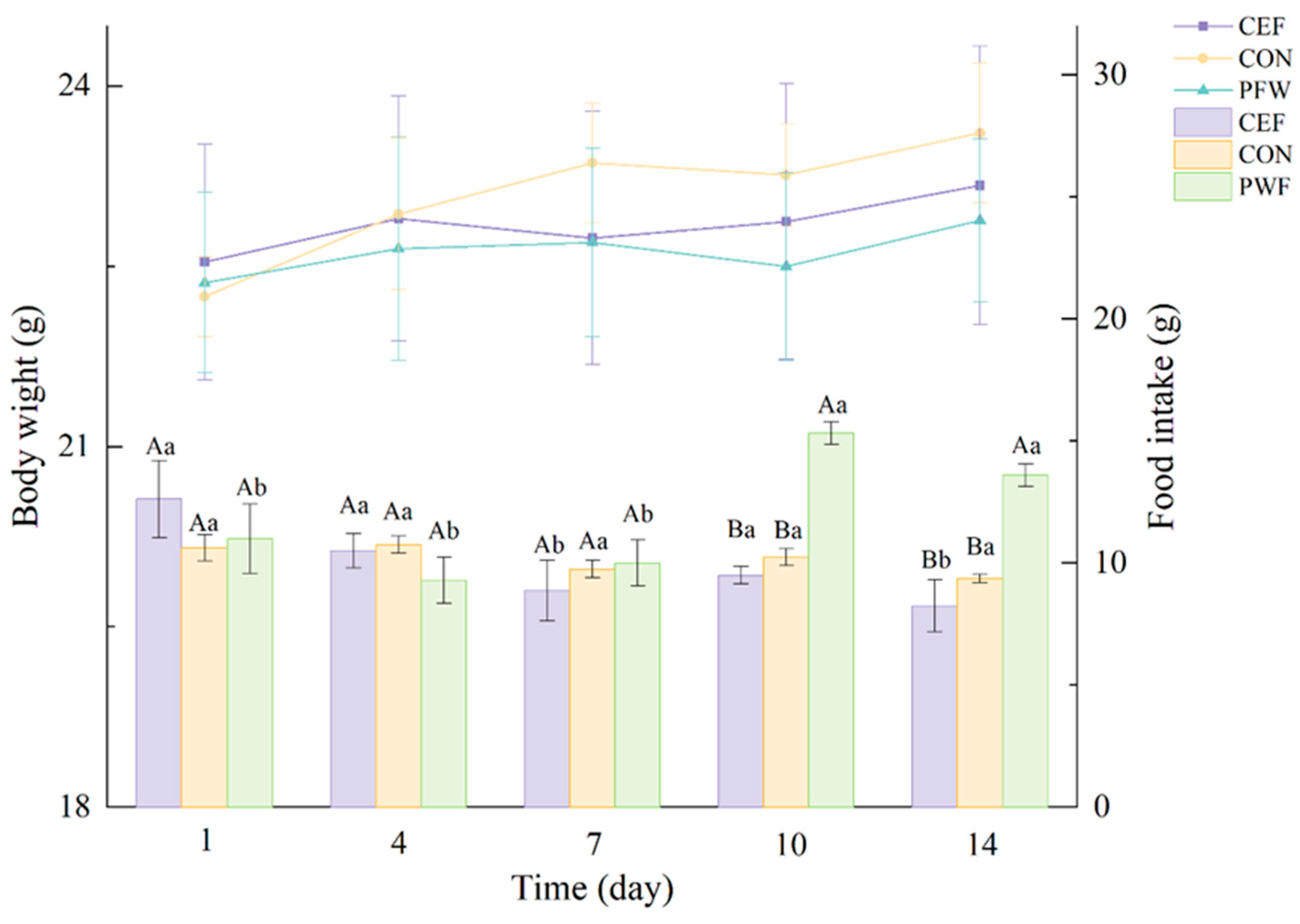
3.2. Effects of PWF on Body Weight and Food Intake by Mice
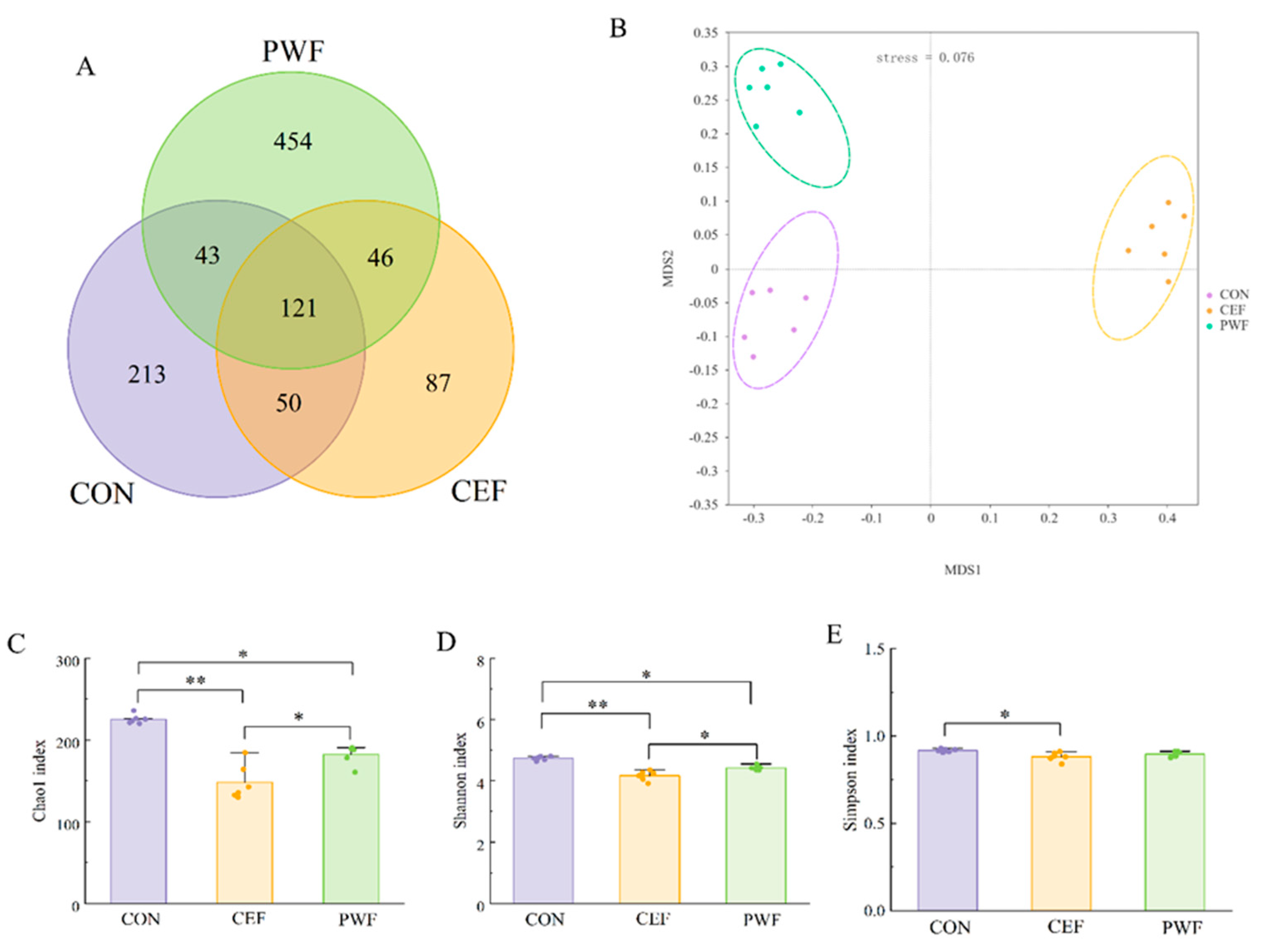
3.3. Effects of PWF on Microbial Community Richness and Diversity
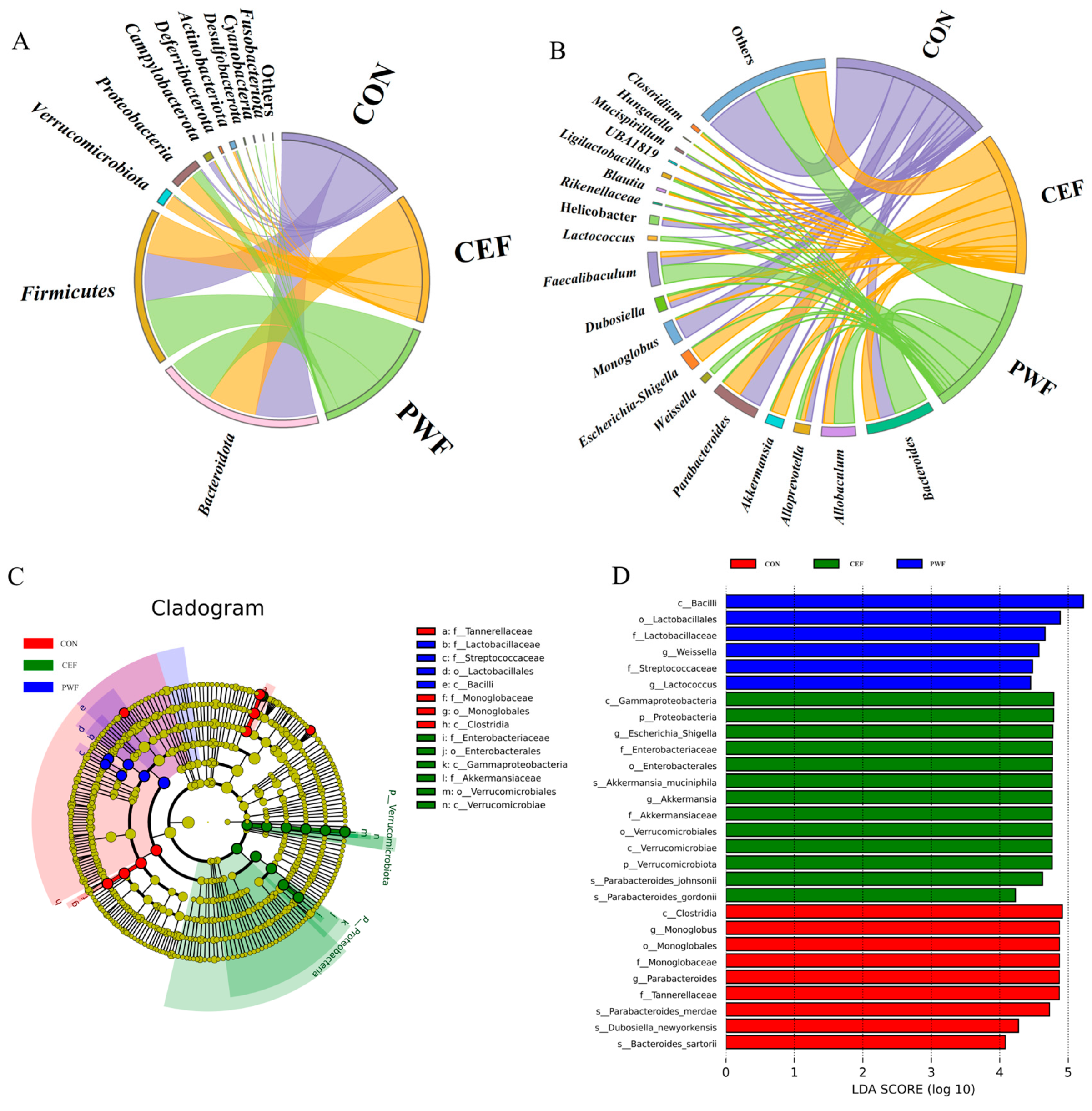
3.4. Effects of PWF on Composition of the Gut Microbial Community
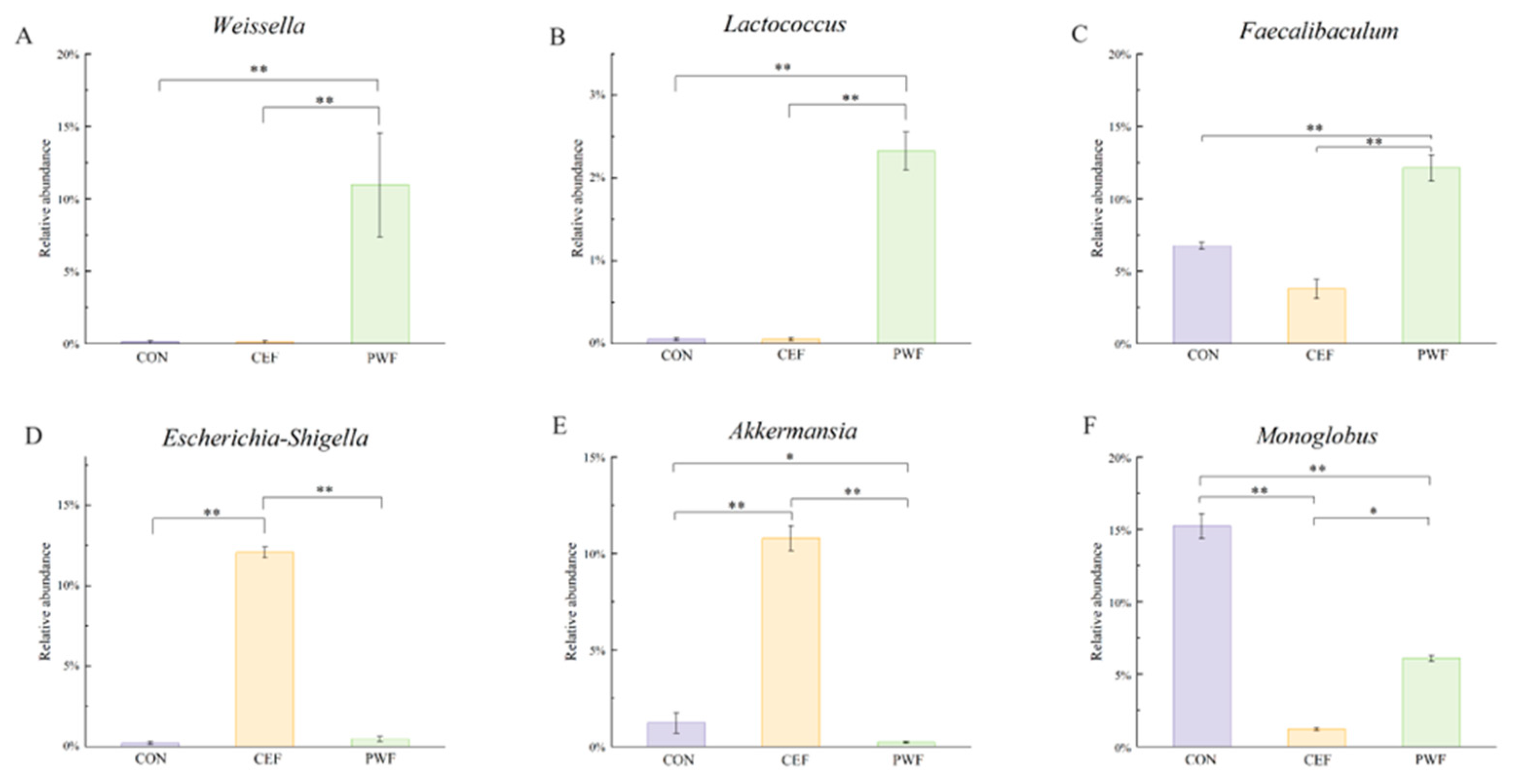
3.5. Analysis of Relative Abundances of Potential Biomarkers in Each Group
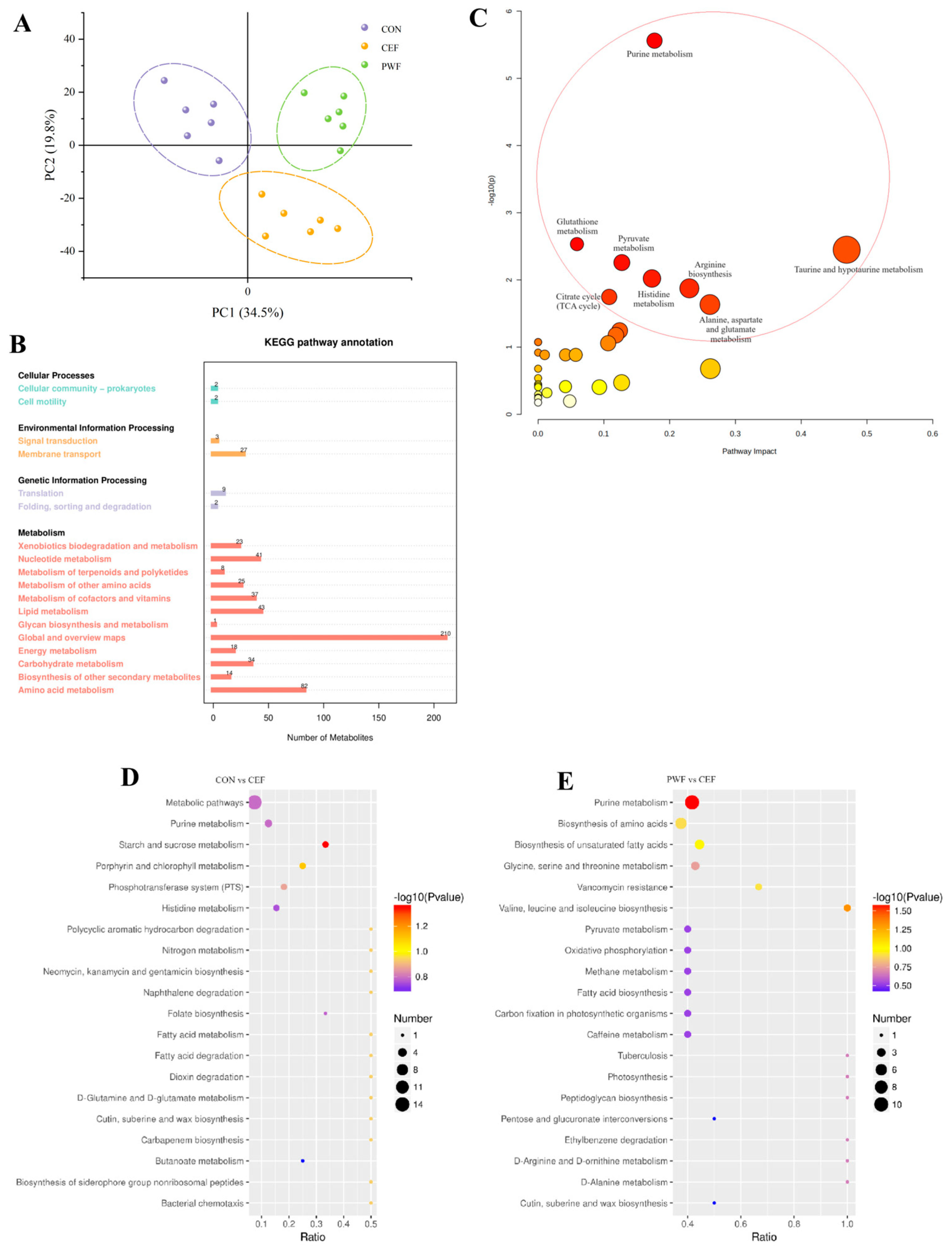
3.6. Effects of PWF on Fecal Metabolomics in Mice
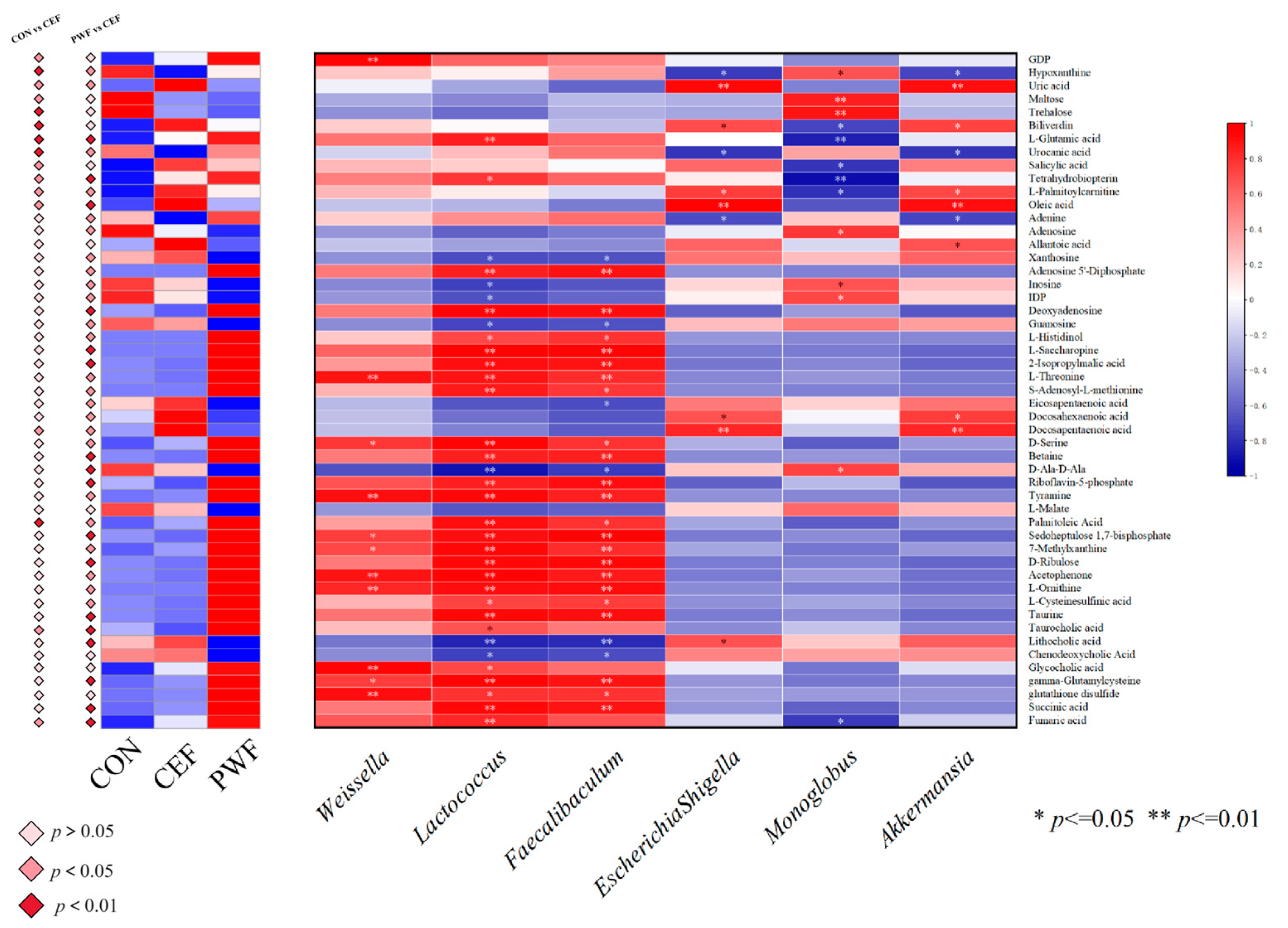
3.7. Correlations between Metabolites of Interest within Divergent Metabolic Pathways and Potential Microorganism Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Gunzburg, J.; Ghozlane, A.; Ducher, A.; Le Chatelier, E.; Duval, X.; Ruppé, E.; Armand-Lefevre, L.; Sablier-Gallis, F.; Burdet, C.; Alavoine, L.; et al. Protection of the Human Gut Microbiome from Antibiotics. J. Infect. Dis. 2018, 217, 628–636. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Yang, H.; Yang, X. Tea polyphenols regulate gut microbiota dysbiosis induced by antibiotic in mice. Food Res. Int. 2021, 141, 110153. [Google Scholar] [CrossRef]
- Dimidi, E.; Cox, S.; Rossi, M.; Whelan, K. Fermented Foods: Definitions and Characteristics, Impact on the Gut Microbiota and Effects on Gastrointestinal Health and Disease. Nutrients 2019, 11, 1806. [Google Scholar] [CrossRef]
- Li, H.; Zhou, D.; Gan, R.; Huang, S.; Zhao, C.; Shang, A.; Xu, X.; Li, H. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef]
- Gryaznova, M.; Smirnova, Y.; Burakova, I.; Syromyatnikov, M.; Chizhkov, P.; Popov, E.; Popov, V. Changes in the Human Gut Microbiome Caused by the Short-Term Impact of Lactic Acid Bacteria Consumption in Healthy People. Probiotics Antimicrob. Proteins. 2023, 24, 86. [Google Scholar] [CrossRef]
- Wu, W.; Chen, T.; Zhao, M.; Feng, Y. Effect of co-inoculation of different halophilic bacteria and yeast on the flavor of fermented soy sauce. Food Biosci. 2023, 51, 102292. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, C.; Qin, X.; Zhou, B.; Liu, X.; Liu, T.; Xie, R.; Liu, J.; Wang, B.; Cao, H. Saccharomyces boulardii, a yeast probiotic, inhibits gut motility through upregulating intestinal serotonin transporter and modulating gut microbiota. Pharmacol. Res. 2022, 181, 106291. [Google Scholar] [CrossRef]
- Luo, J.; Xiao, S.; Wang, J.; Wang, B.; Cai, Y.; Hu, W. The Metabolite Profiling and Microbial Community Dynamics during Pineapple By-Product Fermentation Using Co-Inoculation of Lactic Acid Bacteria and Yeast. Fermentation 2023, 9, 79. [Google Scholar] [CrossRef]
- Luo, J.; Xiao, S.; Wang, B.; Cai, Y.; Wang, J. In vitro fermentation of pineapple–whey protein fermentation product on human intestinal microbiota derived from fecal microbiota transplant donors. LWT 2024, 191, 115637. [Google Scholar] [CrossRef]
- Sireswar, S.; Dey, G.; Biswas, S. Influence of fruit-based beverages on efficacy of Lacticaseibacillus rhamnosus GG (Lactobacillus rhamnosus GG) against DSS-induced intestinal inflammation. Food Res. Int. 2021, 149, 110661. [Google Scholar] [CrossRef]
- Lee, J.; D’Aigle, J.; Atadja, L.; Quaicoe, V.; Honarpisheh, P.; Ganesh, B.P.; Hassan, A.; Graf, J.; Petrosino, J.; Putluri, N.; et al. Gut Microbiota–Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ. Res. 2020, 127, 453–465. [Google Scholar] [CrossRef]
- Yuan, J.; Qin, S.; Hu, S.; Liu, Z.; Song, Y.; Li, L. Restoration of cefixime-induced gut microbiota changes by a prebiotic blend in a mouse model. Appl. Microbiol. Biotechnol. 2022, 106, 5197–5209. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, W.; Li, X.; Yang, H. Interaction between Commensal Bacteria, Immune Response and the Intestinal Barrier in Inflammatory Bowel Disease. Front. Immunol. 2021, 12, 761981. [Google Scholar] [CrossRef]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Wang, M.; Qin, Y.; Liu, Y.; Yang, H.; Wang, J.; Ru, S.; Cui, P. Short-term exposure to enrofloxacin causes hepatic metabolism disorder associated with intestinal flora dysbiosis in adult marine medaka (Oryzias melastigma). Mar. Pollut. Bull. 2023, 192, 114966. [Google Scholar] [CrossRef]
- Yang, X.; Liu, P.; Yu, H.; Ling, M.; Ma, M.; Wang, Q.; Tang, X.; Shen, Z.; Zhang, Y. Comparative analysis of the intestinal flora of BmNPV-resistant and BmNPV-sensitive silkworm varieties. Microb. Pathog. 2024, 191, 106649. [Google Scholar] [CrossRef]
- Xu, G.; Zhao, J.; Shi, K.; Xu, Y.; Hu, H.; Xu, X.; Hu, T.; Zhang, P.; Yao, J.; Pan, S. Trends in valorization of citrus by-products from the net-zero perspective: Green processing innovation combined with applications in emission reduction. Trends Food Sci. Technol. 2023, 137, 124–141. [Google Scholar] [CrossRef]
- Jo, H.E.; Kwon, M.; Whon, T.W.; Kim, D.W.; Yun, M.; Lee, J.; Shin, M.; Kim, S.; Choi, H. Alteration of Gut Microbiota after Antibiotic Exposure in Finishing Swine. Front. Microbiol. 2021, 12, 596002. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Nobel, Y.R.; Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Lightdale, C.J.; Toussaint, N.C.; Abrams, J.A. Increasing Dietary Fiber Intake Is Associated with a Distinct Esophageal Microbiome. Clin. Transl. Gastroenterol. 2018, 9, e199. [Google Scholar] [CrossRef]
- Saleena, L.A.K.; Teo, M.Y.M.; How, Y.H.; In, L.L.A.; Pui, L.P. Immunomodulatory action of Lactococcus lactis. J. Biosci. Bioeng. 2023, 135, 1–9. [Google Scholar] [CrossRef]
- Liang, J.; Li, X.; Lei, W.; Tan, P.; Han, M.; Li, H.; Yue, T.; Wang, Z.; Gao, Z. Serum metabolomics combined with 16S rRNA sequencing to reveal the effects of Lycium barbarum polysaccharide on host metabolism and gut microbiota. Food Res. Int. 2023, 165, 112563. [Google Scholar] [CrossRef]
- Dey, D.K.; Koo, B.G.; Sharma, C.; Kang, S.C. Characterization of Weissella confusa DD_A7 isolated from kimchi. LWT 2019, 111, 663–672. [Google Scholar] [CrossRef]
- Dey, D.K.; Kang, S.C. Weissella confusa DD_A7 pre-treatment to zebrafish larvae ameliorates the inflammation response against Escherichia coli O157:H7. Microbiol. Res. 2020, 237, 126489. [Google Scholar] [CrossRef]
- Lakra, A.K.; Domdi, L.; Hanjon, G.; Tilwani, Y.M.; Arul, V. Some probiotic potential of Weissella confusa MD1 and Weissella cibaria MD2 isolated from fermented batter. LWT 2020, 125, 109261. [Google Scholar] [CrossRef]
- Xie, Y.; Pei, F.; Liu, Y.; Liu, Z.; Chen, X.; Xue, D. Fecal fermentation and high-fat diet-induced obesity mouse model confirmed exopolysaccharide from Weissella cibaria PFY06 can ameliorate obesity by regulating the gut microbiota. Carbohydr. Polym. 2023, 318, 121122. [Google Scholar] [CrossRef]
- Liu, J.; Yu, W.; Wang, C.; Li, S.; Zhang, W. Garlic (Allium sativum) polysaccharides ameliorates hepatic injury and fat accumulation in mice with metabolic associated fatty liver disease (MAFLD). J. Funct. Foods 2022, 99, 105342. [Google Scholar] [CrossRef]
- Ding, J.; Ruan, C.; Guan, Y.; Li, H.; Du, W.; Lu, S.; Wen, X.; Tang, K.; Chen, Y. Nontargeted metabolomic and multigene expression analyses reveal the mechanism of oil biosynthesis in sea buckthorn berry pulp rich in palmitoleic acid. Food Chem. 2022, 374, 131719. [Google Scholar] [CrossRef]
- Abbasi, A.; Bazzaz, S.; Da, C.A.; Khorshidian, N.; Saadat, Y.R.; Sabahi, S.; Ozma, M.A.; Lahouty, M.; Aslani, R.; Mortazavian, A.M. A Critical Review on Akkermansia muciniphila: Functional Mechanisms, Technological Challenges, and Safety Issues. Probiotics Antimicrob. Proteins. 2023, 99, 356–364. [Google Scholar] [CrossRef]
- Morais, V.N.D.; Gomes, M.J.C.; Grancieri, M.; Moreira, L.D.P.D.; Toledo, R.C.L.; Costa, N.M.B.; Da Silva, B.P.; Martino, H.S.D. Chia (Salvia hispanica L.) flour modulates the intestinal microbiota in Wistar rats fed a high-fat and high-fructose diet. Food Res. Int. 2023, 172, 113095. [Google Scholar] [CrossRef]
- Kasinathan, N.K.; Subramaniya, B.; Sivasithamparam, N.D. NF-κB/twist mediated regulation of colonic inflammation by lupeol in abating dextran sodium sulfate induced colitis in mice. J. Funct. Foods. 2018, 41, 240–249. [Google Scholar] [CrossRef]
- Wu, T.; Shen, M.; Yu, Q.; Chen, Y.; Chen, X.; Yang, J.; Huang, L.; Guo, X.; Xie, J. Cyclocarya paliurus polysaccharide improves metabolic function of gut microbiota by regulating short-chain fatty acids and gut microbiota composition. Food Res. Int. 2021, 141, 110119. [Google Scholar] [CrossRef]
- Li, Y.; Shi, J.; Wu, H.; Wong, K.; Cheung, P.C.K.; Lv, X.; Yang, Q.; Zhang, B. Effects of pomelo sponge fermented by Monascus ruber on cholesterol metabolism and intestinal microbiota in high-fat diet mice. Food Biosci. 2023, 56, 103376. [Google Scholar] [CrossRef]
- Hu, B.; Liu, C.; Jiang, W.; Zhu, H.; Zhang, H.; Qian, H.; Zhang, W. Chronic in vitro fermentation and in vivo metabolism: Extracellular polysaccharides from Sporidiobolus pararoseus regulate the intestinal microbiome of humans and mice. Int. J. Biol. Macromol. 2021, 192, 398–406. [Google Scholar] [CrossRef]
- AL Mughram, M.H.; Catalano, C.; Herrington, N.B.; Safo, M.K.; Kellogg, G.E. 3D interaction homology: The hydrophobic residues alanine, isoleucine, leucine, proline and valine play different structural roles in soluble and membrane proteins. Front. Mol. Biosci. 2023, 10, 1116868. [Google Scholar] [CrossRef]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 2018, 15, 33. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, G.; Yun, S.H.; Lee, W.; Yu, J.; Kim, S.K.; Lee, B. The role of SHMT2 in modulating lipid metabolism in hepatocytes via glycine-mediated mTOR activation. Amino Acids. 2022, 54, 823–834. [Google Scholar] [CrossRef]
- Li, M.; Wu, Y.; Ye, L. The Role of Amino Acids in Endothelial Biology and Function. Cells 2022, 11, 1372. [Google Scholar] [CrossRef]
- Yu, J.; Sun, J.; Sun, M.; Li, W.; Qi, D.; Zhang, Y.; Han, C. Protective mechanism of Coprinus comatus polysaccharide on acute alcoholic liver injury in mice, the metabolomics and gut microbiota investigation. Food Sci. Hum. Wellness 2024, 13, 401–413. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Xiao, S.; Ma, D.; Xiang, J.; Wang, B.; Cai, Y.; Wang, J. Investigating the Impact of Pineapple–Whey Protein Fermentation Products on Cefixime-Induced Intestinal Flora Dysbiosis in Mice Using 16S Sequencing and Untargeted Metabolomics Techniques. Foods 2024, 13, 1927. https://doi.org/10.3390/foods13121927
Luo J, Xiao S, Ma D, Xiang J, Wang B, Cai Y, Wang J. Investigating the Impact of Pineapple–Whey Protein Fermentation Products on Cefixime-Induced Intestinal Flora Dysbiosis in Mice Using 16S Sequencing and Untargeted Metabolomics Techniques. Foods. 2024; 13(12):1927. https://doi.org/10.3390/foods13121927
Chicago/Turabian StyleLuo, Jiawei, Shan Xiao, Da Ma, Junhan Xiang, Bo Wang, Yanxue Cai, and Jihui Wang. 2024. "Investigating the Impact of Pineapple–Whey Protein Fermentation Products on Cefixime-Induced Intestinal Flora Dysbiosis in Mice Using 16S Sequencing and Untargeted Metabolomics Techniques" Foods 13, no. 12: 1927. https://doi.org/10.3390/foods13121927