
Proteomic Analysis of the Characteristic Flavor Components in Bacillus subtilis BSNK-5-Fermented Soymilk

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Fermented Soymilk
2.3. Protein Extraction
2.4. Protein Molecular Weight Analysis using Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE)
2.5. Proteolytic Enzymolysis
2.6. Protein Labeling
2.7. Protein Fractionation
2.8. Peptide Profiling Analysis
2.9. Bioinformatics Analysis
3. Results
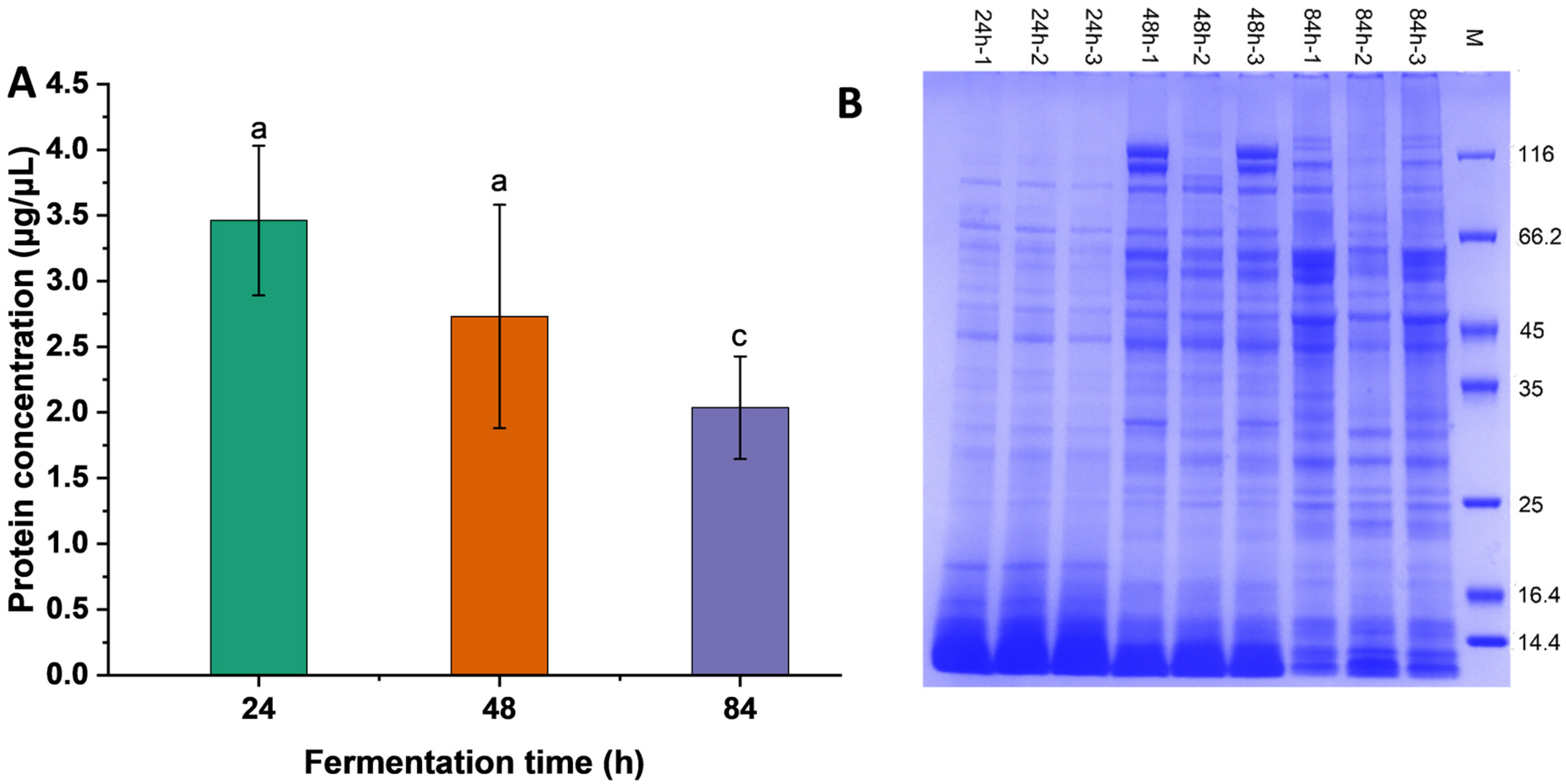
3.1. Protein Concentration and Molecular Weight Distribution
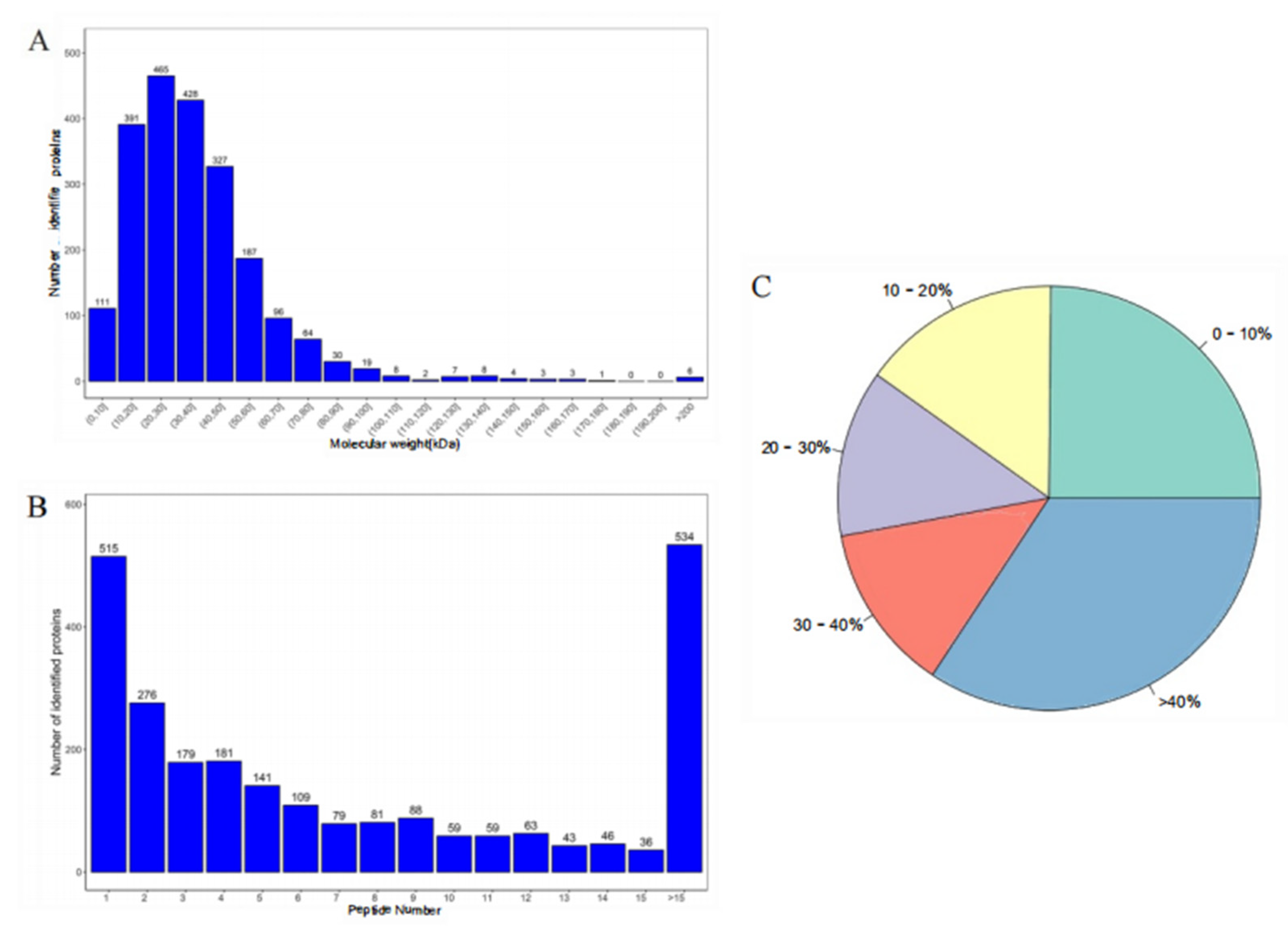
3.2. Identification of Proteins using Quantitative Proteomics Analysis
3.3. Protein Identification
3.4. Global Analysis of Differentially Expressed Proteins
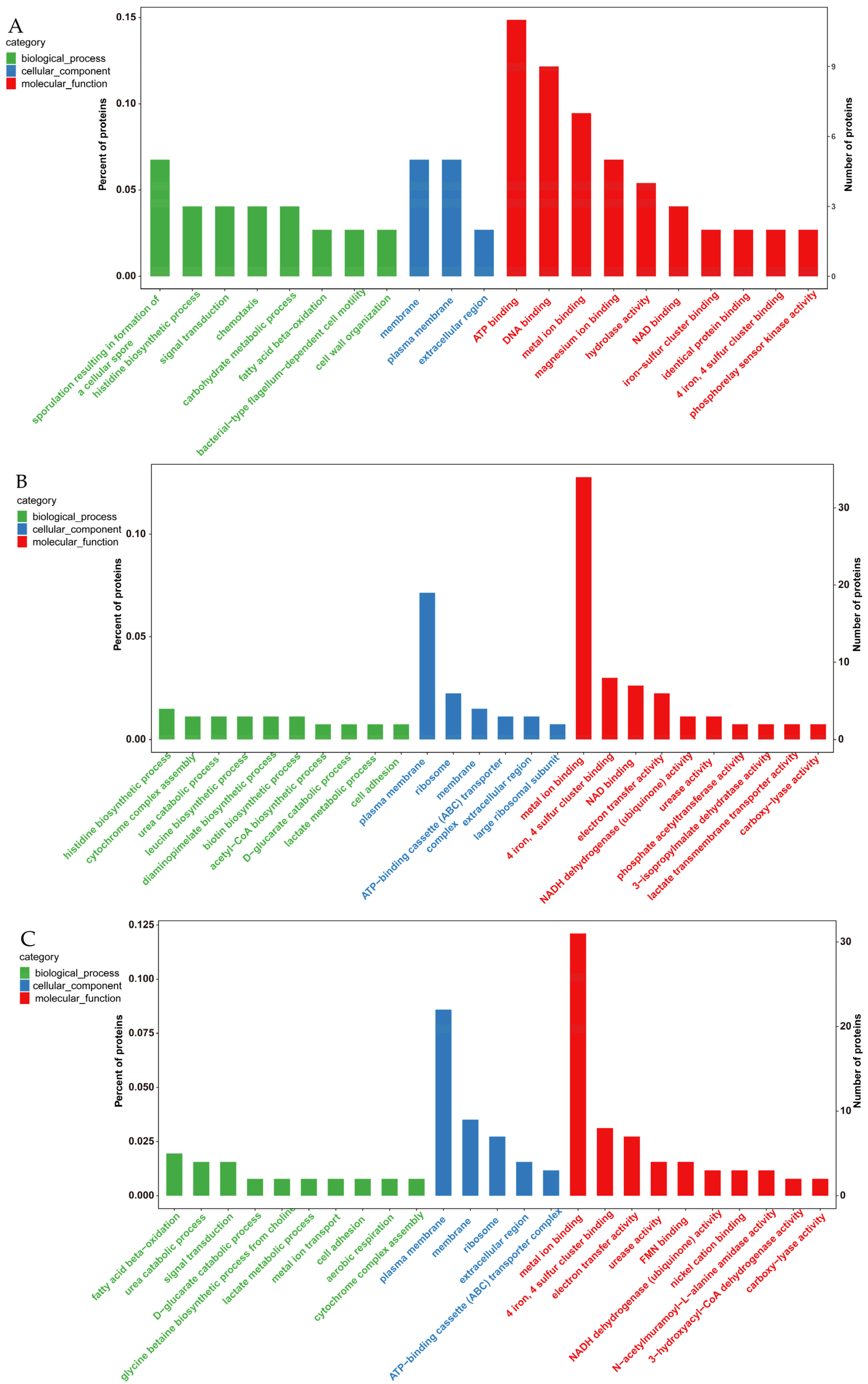
3.4.1. GO Enrichment Analysis of Differentially Expressed Proteins
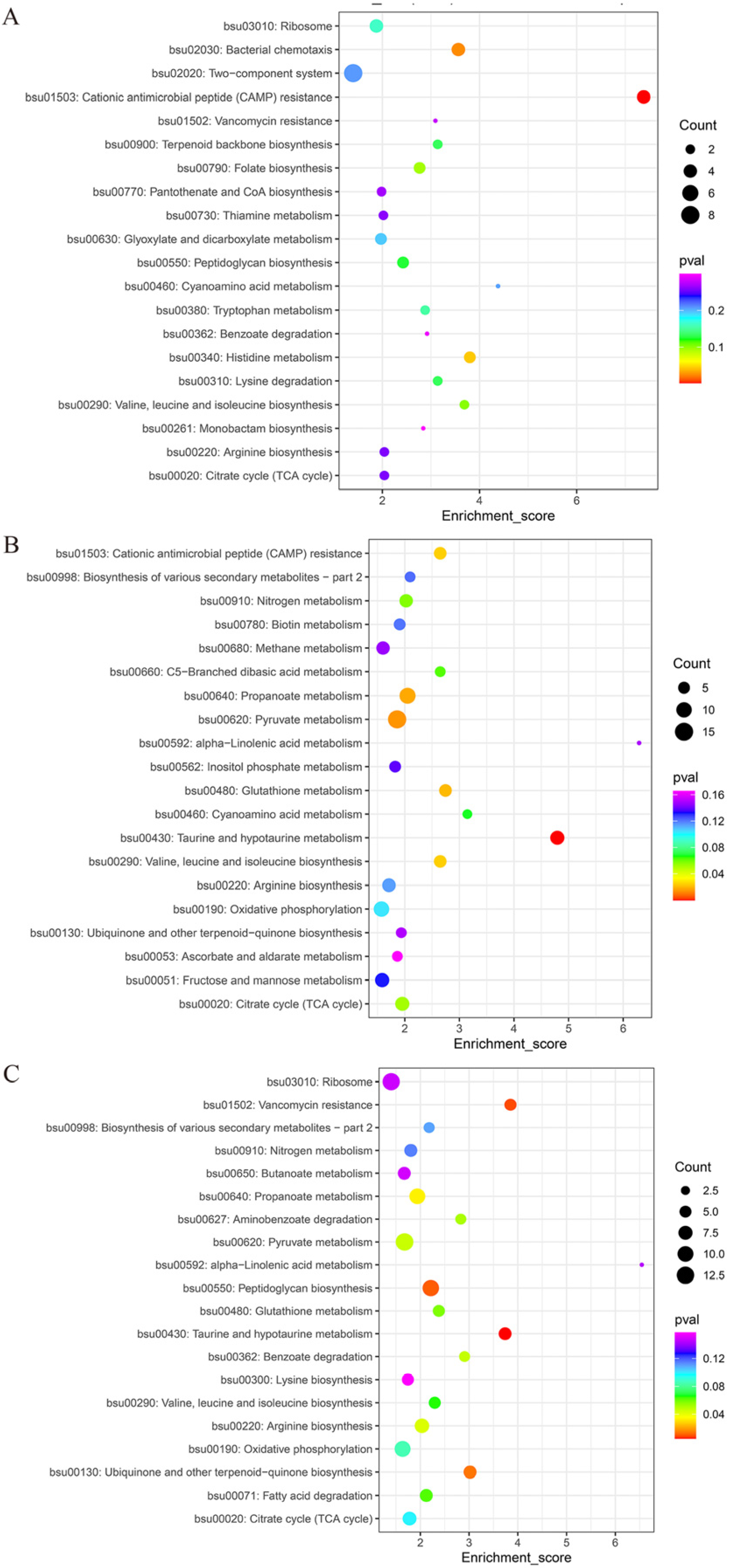
3.4.2. KEGG Analysis of Differential Proteins
3.5. Selection of Differentially Expressed Proteins Associated with Characteristic Flavor Formation
4. Discussion
4.1. Metabolic Pathways of Amino Acids
4.2. Metabolic Pathways of Pyrazine Compounds
4.3. Metabolic Pathways of BCFAs and Acetic Acid
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhan, Q.; Thakur, K.; Feng, J.-Y.; Zhu, Y.-Y.; Zhang, J.-G.; Wei, Z.-J. LC–MS based metabolomics analysis of okara fermented by Bacillus subtilis DC-15: Insights into nutritional and functional profile. Food Chem. 2023, 413, 135656. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, Y.; Zhao, A.; Wu, Y.; Zhang, Y.; Yang, X. Quantitative analyses for several nutrients and volatile components during fermentation of soybean by Bacillus subtilis. Food Chem. 2022, 374, 131725. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Ikehara, S.; Kamiya, K.; Kajita, E.; Sato, Y.; Kouda, K.; Tamaki, J.; Kagamimori, S.; Iki, M. Natto Intake is Inversely Associated with Osteoporotic Fracture Risk in Postmenopausal Japanese Women. J. Nutr. 2020, 150, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-Y.; Liu, L.-H.; Chen, C.-Y.; Lin, I.F.; Ali, D.; Yueh-Luen Lee, A.; David Wang, H.-M. Bifunctional mechanisms of autophagy and apoptosis regulations in melanoma from Bacillus subtilis fermentation extract. Food Chem. Toxicol. 2021, 150, 112020. [Google Scholar] [CrossRef] [PubMed]
- Jamali, N.; Vahedi, F.; Soltani Fard, E.; Taheri-Anganeh, M.; Taghvimi, S.; Khatami, S.H.; Ghasemi, H.; Movahedpour, A. Nattokinase: Structure, applications and sources. Biocatal. Agric. Biotechnol. 2023, 47, 102564. [Google Scholar] [CrossRef]
- Chiu, H.-W.; Chou, C.-L.; Lee, K.-T.; Shih, C.-C.; Huang, T.-H.; Sung, L.-C. Nattokinase attenuates endothelial inflammation through the activation of SRF and THBS1. Int. J. Biol. Macromol. 2024, 268, 131779. [Google Scholar] [CrossRef] [PubMed]
- Li, D.F.; Hou, L.Z.; Hu, M.; Gao, Y.X.; Tian, Z.L.; Fan, B.; Li, S.Y.; Wang, F.Z. Recent Advances in Nattokinase-Enriched Fermented Soybean Foods: A Review. Foods 2022, 11, 1867. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.X.; Xu, B.; Fan, H.R.; Zhang, M.R.; Zhang, L.J.; Lu, C.; Zhang, N.N.; Fan, B.; Wang, F.Z.; Li, S. 1H NMR-based chemometric metabolomics characterization of soymilk fermented by Bacillus subtilis BSNK-5. Food Res. Int. 2020, 138, 109686. [Google Scholar] [CrossRef]
- Chukeatirote, E. Thua nao: Thai fermented soybean. J. Ethn. Foods 2015, 2, 115–118. [Google Scholar] [CrossRef]
- Liu, Y.; Song, H.; Luo, H. Correlation between the key aroma compounds and gDNA copies of Bacillus during fermentation and maturation of natto. Food Res. Int. 2018, 112, 175–183. [Google Scholar] [CrossRef]
- Riley, S.; Lale, A.; Nguyen, V.; Xi, H.; Wilkinson, K.; Searle, I.R.; Fisk, I. Volatile profiles of commercial vetch prepared via different processing methods. Food Chem. 2022, 395, 133569. [Google Scholar] [CrossRef] [PubMed]
- Keong, L.Y.E.; Toh, M.; Lu, Y.; Liu, S.-Q. Biotransformation of okara (soybean residue) through solid-state fermentation using probiotic Bacillus subtilis and Bacillus coagulans. Food Biosci. 2023, 55, 103056. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.-l.; Xu, P.; Guo, S.; Zhou, T.; Li, N. Soy protein degradation drives diversity of amino-containing compounds via Bacillus subtilis fermentation. Food Chem. 2022, 388, 133034. [Google Scholar] [CrossRef] [PubMed]
- Kada, S.; Yabusaki, M.; Kaga, T.; Ashida, H.; Yoshida, K.-I. Identification of Two Major Ammonia-Releasing Reactions Involved in Secondary Natto Fermentation. Biosci. Biotechnol. Biochem. 2008, 72, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Mengqi, Y.; Yuxin, Y.; Yingxin, Q.; Riakang, S.; Xinyang, R.; Guang, L. Tandem mass spectrometry tag proteomic analysis of milk and colostrum of Guanzhong dairy goat. Int. Dairy J. 2024, 148, 105781. [Google Scholar] [CrossRef]
- Yang, Z.; Hou, Y.; Zhang, M.; Hou, P.; Liu, C.; Dou, L.; Chen, X.; Zhao, L.; Su, L.; Jin, Y. Unraveling proteome changes of Sunit lamb meat in different feeding regimes and its relationship to flavor analyzed by TMT-labeled quantitative proteomic. Food Chem. 2024, 437, 137657. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, X.; Pan, D.; Xia, Q.; Sun, Y.; Geng, F.; Cao, J. TMT-labeled quantitative proteomic reveals the mechanism of proteolysis and taste improvement of dry-cured bacon with Staphylococcus co-inoculation. Food Chem. 2024, 436, 137711. [Google Scholar] [CrossRef]
- Verkempinck, S.H.E.; Duijsens, D.; Mukherjee, A.; Wilde, P.J. Pea protein extraction method impacts the protein (micro)structural organisation and in vitro digestion kinetics. Food Funct. 2024, 15, 953–966. [Google Scholar] [CrossRef]
- Shi, L.; Pico, J.; Zamani, S.; Castellarin, S.D.; Dee, D.R. Fibrillization of lentil proteins is impacted by the protein extraction conditions and co-extracted phenolics. Food Chem. 2024, 448, 139104. [Google Scholar] [CrossRef]
- Tang, J.; Zhu, X.; Dong, G.; Hannon, S.; Santos, H.M.; Sun, D.-W.; Tiwari, B.K. Comparative studies on enhancing pea protein extraction recovery rates and structural integrity using ultrasonic and hydrodynamic cavitation technologies. LWT 2024, 200, 116130. [Google Scholar] [CrossRef]
- Mansuri, M.S.; Bathla, S.; Lam, T.T.; Nairn, A.C.; Williams, K.R. Optimal conditions for carrying out trypsin digestions on complex proteomes: From bulk samples to single cells. J. Proteom. 2024, 297, 105109. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Bermúdez, I.C.; Jain, D.; Ravindran, A.; Chen, C.W.; Hsu, C.C.; Schmidt, W. Tandem Mass Tag-Based Phosphoproteomics in Plants. Methods Mol. Biol. 2023, 2581, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, Z.; Cupp-Sutton, K.A.; Guo, Y.; Kou, Q.; Smith, K.; Liu, X.; Wu, S. Quantitative Top-Down Proteomics in Complex Samples Using Protein-Level Tandem Mass Tag Labeling. J. Am. Soc. Mass Spectrom. 2021, 32, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yin, X.; Wei, H.; Wang, Z.; Tang, H.; Qiu, Y.; Hao, Y.; Zhang, X.; Bi, H.; Guo, D. Quantitative proteomic analysis of rat retina with experimental autoimmune uveitis based on tandem mass tag (TMT) peptide labeling coupled with LC-MS/MS. J. Chromatogr. B 2020, 1153, 122293. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, C.D.; Aizikov, K.; Batth, T.S.; Kreutzman, A.; Grinfeld, D.; Lange, O.; Mourad, D.; Makarov, A.A.; Olsen, J.V. Limits for Resolving Isobaric Tandem Mass Tag Reporter Ions Using Phase-Constrained Spectrum Deconvolution. J. Proteome Res. 2018, 17, 4008–4016. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Hong, H.; Yang, Q.; Liu, X.; Zhuang, S.; Li, Y.; Liu, J.; Luo, Y. TMT-based proteomic analysis of the fish-borne spoiler Pseudomonas psychrophila subjected to chitosan oligosaccharides in fish juice system. Food Microbiol. 2020, 90, 103494. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Magasanik, B. Role of NAD-linked glutamate dehydrogenase in nitrogen metabolism in Saccharomyces cerevisiae. J. Bacteriol. 1990, 172, 4927–4935. [Google Scholar] [CrossRef] [PubMed]
- Satomura, T.; Shimura, D.; Asai, K.; Sadaie, Y.; Hirooka, K.; Fujita, Y. Enhancement of Glutamine Utilization in Bacillus subtilis through the GlnK-GlnL Two-Component Regulatory System. J. Bacteriol. 2005, 187, 4813–4821. [Google Scholar] [CrossRef]
- Riethdorf, S.; Völker, U.; Gerth, U.; Winkler, A.; Engelmann, S.; Hecker, M. Cloning, nucleotide sequence, and expression of the Bacillus subtilis lon gene. J. Bacteriol. 1994, 176, 6518–6527. [Google Scholar] [CrossRef]
- Cruz-Ramos, H.; Glaser, P.; Wray, L.V.; Fisher, S.H. The Bacillus subtilis ureABC operon. J. Bacteriol. 1997, 179, 3371–3373. [Google Scholar] [CrossRef] [PubMed]
- Commichau, F.M.; Stülke, J. Trigger enzymes: Bifunctional proteins active in metabolism and in controlling gene expression. Mol. Microbiol. 2008, 67, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Wang, Q.; Wei, X.; Ma, X.; Chen, S. Glutamate dehydrogenase (RocG) in Bacillus licheniformis WX-02: Enzymatic properties and specific functions in glutamic acid synthesis for poly-γ-glutamic acid production. Enzym. Microb. Technol. 2017, 99, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Gunka, K.; Newman, J.A.; Commichau, F.M.; Herzberg, C.; Rodrigues, C.; Hewitt, L.; Lewis, R.J.; Stülke, J. Functional Dissection of a Trigger Enzyme: Mutations of the Bacillus subtilis Glutamate Dehydrogenase RocG That Affect Differentially Its Catalytic Activity and Regulatory Properties. J. Mol. Biol. 2010, 400, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Gunka, K.; Commichau, F.M. Control of glutamate homeostasis in Bacillus subtilis: A complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol. Microbiol. 2012, 85, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cao, Y.; Tong, J.; Xu, Y. An Alkylpyrazine Synthesis Mechanism Involving l-Threonine-3-Dehydrogenase Describes the Production of 2,5-Dimethylpyrazine and 2,3,5-Trimethylpyrazine by Bacillus subtilis. Appl. Environ. Microbiol. 2019, 85, e01807–e01819. [Google Scholar] [CrossRef] [PubMed]
- Larroche, C.; Besson, I.; Gros, J.-B. High pyrazine production by Bacillus subtilis in solid substrate fermentation on ground soybeans. Process Biochem. 1999, 34, 667–674. [Google Scholar] [CrossRef]
- Takemura, H.; Ando, N.; Tsukamoto, Y. Breeding of Branched Short-Chain Fatty Acids Non-Producing Natto Bacteria and its Application to Production of Natto with Light Smells. Nippon. Shokuhin Kagaku Kogaku Kaishi 2000, 47, 773–779. [Google Scholar] [CrossRef]
- Hong, C.; Chen, Y.; Li, L.; Chen, S.; Wei, X. Identification of a Key Gene Involved in Branched-Chain Short Fatty Acids Formation in Natto by Transcriptional Analysis and Enzymatic Characterization in Bacillus subtilis. J. Agric. Food Chem. 2017, 65, 1592–1597. [Google Scholar] [CrossRef]
- Sirobhushanam, S.; Galva, C.; Sen, S.; Wilkinson, B.J.; Gatto, C. Broad substrate specificity of phosphotransbutyrylase from Listeria monocytogenes: A potential participant in an alternative pathway for provision of acyl CoA precursors for fatty acid biosynthesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 1102–1110. [Google Scholar] [CrossRef]
- Oku, H.; Kaneda, T. Biosynthesis of branched-chain fatty acids in Bacillus subtilis. A decarboxylase is essential for branched-chain fatty acid synthetase. J. Biol. Chem. 1988, 263, 18386–18396. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Akita, K.; Sakai, R.; Shimomura, Y. Analysis of branched-chain α-keto acid dehydrogenase complex activity in rat tissues using α-keto [1-13C]isocaproate as substrate. Anal. Biochem. 2010, 399, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, W.-X.; Fu, J.; Zhang, B.; Tang, Y.-J. Engineering Bacillus subtilis for acetoin production from glucose and xylose mixtures. J. Biotechnol. 2013, 168, 499–505. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Process | Accession | Gene Name | Protein Description | Group 1 FC | Group 2 FC | Group 3 FC |
|---|---|---|---|---|---|---|
| Alanine, aspartate, and glutamate metabolism | A0A6H0H898 | pruA | Adenylosuccinate synthetase | 0.17 | 0.12 | |
| G4EZA6 | purF | Amidophosphoribosyl transferase | 0.38 | 0.33 | ||
| A0A6H2KH29 | ald | Alanine dehydrogenase | 0.34 | 0.23 | ||
| P94427 | gabT | Probable 4-aminobutyrate aminotransferase | 0.14 | 0.12 | ||
| A0A199WDG9 | rocG | Glutamate dehydrogenase | 0.19 | 0.17 | ||
| A0A6H2K2D7 | gltA | Glutamate synthase (NADPH) large chain | 0.37 | 0.40 | ||
| A0A6H2K2B8 | gltB | NADPH small chain | 0.49 | |||
| A0A1B2AVT2 | carB | Carbamoyl-phosphate synthase large chain | 0.20 | 0.21 | ||
| Valine, leucine, and isoleucine degradation | A0A6I4DBI8 | bfmBB | Branched-chain alpha-keto dehydrogenase complex dihydrolipoyllysine-residue (2-methylpropanoyl) transferase | 2.29 | 2.02 | |
| A0A6H2KKS5 | iolA | Methylmalonate/inositol-semialdehyde dehydrogenase | 0.44 | 0.49 | ||
| G4EYT5 | BSSC8_35150 | Dihydrolipoyl dehydrogenase | 0.13 | 0.17 | ||
| A0A6H0H9I7 | HCN55_12915 | BSU23920 | 0.23 | 0.19 | ||
| A0A6H0H6V2 | HCN55_17715 | Acetyl-CoA C-acyltransferase | 0.39 | 0.23 | ||
| Valine, leucine, and isoleucine biosynthesis | P37253 | ilvC | Ketol-acidreductoisomerase (NADP(+)) | 0.43 | 0.10 | 0.22 |
| A0A6I4D8C8 | ilvD | Dihydroxy-acid dehydratase | 0.41 | 0.45 | ||
| P05645 | leuB | 3-isopropylmalate dehydrogenase | 0.35 | 0.15 | 0.42 | |
| G4EST7 | leuC | 3-isopropylmalate dehydratase large subunit | 0.21 | 0.35 | ||
| P94568 | leuD | 3-isopropylmalate dehydratase small subunit | 0.30 | |||
| A0A6H0H8M1 | alsS | Acetolactate synthase AlsS | 0.32 | 0.27 | ||
| Glycine, serine, and threonine metabolism | Q04797 | asd | Aspartate-semialdehyde dehydrogenase | 0.45 | ||
| A0A1B2B4F9 | kbl | 8-amino-7-ketopelargonate synthase | 0.47 | 0.34 | ||
| O31776 | tdh | L-threonine 3-dehydrogenase | 0.20 | 0.20 | ||
| A0A6H2K6M6 | yvcT | Glyoxylate reductase YvcT | 0.44 | |||
| P08495 | lysC | Aspartokinase 2 | 0.26 | 0.41 | ||
| A0A199WF35 | gbsA | Betaine aldehyde dehydrogenase | 2.46 | |||
| P71017 | gbsB | Choline dehydrogenase | 2.70 | 2.64 | ||
| A3F3S8 | trpA | Tryptophan synthase alpha chain | 2.11 | |||
| A0A6H2K323 | trpB | Tryptophan synthase beta chain | 2.60 | 3.69 | ||
| Arginine and proline metabolism | A0A6H0H1Z3 | proA | Glutamate-5-semialdehyde dehydrogenase | 0.37 | 0.29 | |
| P21885 | speA | Arginine decarboxylase | 0.31 | 0.36 | ||
| G4EP73 | rocD | Ornithine aminotransferase | 0.27 | 0.26 | ||
| P39138 | rocF | Arginase | 0.47 | |||
| Arginine biosynthesis | A0A6H2KBK9 | argC | N-acetyl-gamma-glutamyl-phosphate reductase | 0.27 | 0.40 | |
| A0A6H2K084 | argF | Ornithine carbamoyltransferase | 0.12 | 0.09 | ||
| O34984 | yodQ | Uncharacterized metallohydrolase YodQ | 0.27 | 0.39 | 1.44 | |
| P77837 | ureC | Urease subunit alpha | 0.14 | 0.17 | ||
| P71035 | ureB | Urease subunit beta | 0.18 | 0.18 | ||
| P75030 | ureA | Urease subunit gamma | 0.07 | 0.12 | ||
| G4EQB1 | BSSC8_05410 | Urea amidohydrolase | 2.77 | 0.20 | ||
| Purine metabolism | A0A6H0GZI4 | HCN55_03695 | Adenine deaminase | 0.46 | 0.27 | |
| G4EWI5 | sat | Sulfate adenylyltransferase | 0.49 | 0.18 | 0.38 | |
| A0A6H2JX42 | dgk | Deoxyguanosine kinase DGK | 0.38 | |||
| A0A1B2B5S8 | rdgB | dITP/XTP pyrophosphatase | 0.36 | |||
| A0A6H2K5V9 | pucH | Allantoinase | 0.16 | 0.18 | ||
| O32141 | pucL | Uric acid degradation bifunctional protein PucL | 0.31 | 0.45 | ||
| A0A6H2KDK2 | nrdEB | Ribonucleotide reductase of class Ib (Aerobic) | 2.04 | 1.79 | ||
| Pentose phosphate pathway | A0A6H0GYQ1 | HCN55_02205 | SDR family oxidoreductase | 0.39 | 0.93 | 2.34 |
| A0A6H2KGT5 | zwf | NADP-dependent glucose-6-phosphate 1-dehydrogenase | 0.34 | 0.45 | ||
| G4EU42 | BSSC8_18720 | 6-phosphogluconate dehydrogenase, decarboxylating | 0.0 | 0.35 | ||
| Q03224 | glpX | Fructose-1,6-bisphosphatase class 2 | 2.06 | 1.81 | ||
| TCA cycle | A0A1B2BE85 | pckA | Phosphoenolpyruvate carboxykinase (ATP) | 2.34 | 2.23 | |
| A0A6H0H2Z7 | odhB | 2-oxoglutarate dehydrogenase complex dihydrolipoyllysine-residue succinyltransferase | 2.37 | 3.70 | ||
| A0A6H0H5Z3 | sdhA | Succinate dehydrogenase flavoprotein subunit | 0.42 | 0.37 | ||
| A0A6H2K2M0 | pycA | Pyruvate carboxylase | 2.20 | 2.47 | ||
| A0A6I4DHN9 | mmgD | 2-methylcitrate synthase | 0.49 | |||
| G4ESR9 | BSSC8_13990 | Succinate dehydrogenase (quinone) | 0.43 | 0.43 | ||
| G4EWR2 | BSSC8_27920 | Pyruvate carboxylase | 2.03 | 2.09 | ||
| Pyruvate metabolism | A0A199WLB6 | ackA | Acetate kinase | 0.45 | 0.49 | |
| A0A6H0H0J6 | acoC | Acetoin dehydrogenase complex dihydrolipoyllysine-residue acetyltransferase | 0.11 | 0.08 | ||
| A0A6H0H4J8 | accC | Acetyl-CoA carboxylase biotin carboxylase subunit | 2.16 | 2.25 | ||
| A0A6H0H6N9 | acsA | Acetate--CoA ligase | 2.24 | 2.59 | ||
| G4EQ01 | BSSC8_04310 | Phosphotransacetylase | 0.41 | 0.42 | ||
| G4EQ65 | BSSC8_04950 | Malate dehydrogenase (oxaloacetate-decarboxylating) | 0.35 | 0.31 | ||
| G4EQR2 | BSSC8_06920 | Putative phosphotransferase | 2.04 | 3.04 | ||
| G4F094 | ldh | L-lactate dehydrogenase | 0.09 | 0.12 | ||
| G9LQ56 | yngH | Acetyl-CoA carboxylase biotin carboxylase subunit | 0.46 | |||
| O35031 | acyP | Acylphosphatase | 0.43 | |||
| P39646 | pta | Phosphate acetyltransferase | 0.37 | |||
| P54501 | yqgX | Probable metallo-hydrolase YqgX | 0.39 | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Wang, J.; Gao, Y.; Fan, B.; Wang, F.; Li, S. Proteomic Analysis of the Characteristic Flavor Components in Bacillus subtilis BSNK-5-Fermented Soymilk. Foods 2024, 13, 2399. https://doi.org/10.3390/foods13152399
Hu M, Wang J, Gao Y, Fan B, Wang F, Li S. Proteomic Analysis of the Characteristic Flavor Components in Bacillus subtilis BSNK-5-Fermented Soymilk. Foods. 2024; 13(15):2399. https://doi.org/10.3390/foods13152399
Chicago/Turabian StyleHu, Miao, Jiao Wang, Yaxin Gao, Bei Fan, Fengzhong Wang, and Shuying Li. 2024. "Proteomic Analysis of the Characteristic Flavor Components in Bacillus subtilis BSNK-5-Fermented Soymilk" Foods 13, no. 15: 2399. https://doi.org/10.3390/foods13152399
APA StyleHu, M., Wang, J., Gao, Y., Fan, B., Wang, F., & Li, S. (2024). Proteomic Analysis of the Characteristic Flavor Components in Bacillus subtilis BSNK-5-Fermented Soymilk. Foods, 13(15), 2399. https://doi.org/10.3390/foods13152399