
Transcriptome Analysis Reveals the Variations in Enzyme Production of Saccharopolyspora rosea A22 under Different Temperatures

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Materials
2.2. Cultivation and Morphological Observation of Saccharopolyspora rosea A22
2.3. Measurement of Biomass
2.4. Production of Cooked Wheat Qu
- (1)
- Wheat pre-treatment: High-quality red skin wheat was selected to make wheat Qu and the wheat grains were kept dry. The wheat was screened at the conveyor port of the wheat mill to remove impurities and ensure cleanliness. It was then crushed using a wheat mill, with the roller gap set at 1.7–1.8 mm, resulting in 2 to 3 pieces of wheat per grain to break the wheat peel and expose the starch in the wheat grain.
- (2)
- Moistening material cooking: Approximately 40% of normal temperature pure water (by total mass) was added to moisten the material. Care was taken to add water and wheat in batches to avoid local areas becoming too wet or too dry. The mixture was then placed into a pre-heated boiling steam box and steamed for about 40 min.
- (3)
- Cooling inoculation: The digester was opened, and the material was discharged to reduce the temperature to 45 °C and remove any caking material. All cultures were mixed well and an appropriate volume of cultures (5% of the total mass) was weighed using a clean measuring cylinder and evenly added to the material.
- (4)
- Wheat Qu culture: The mixed wheat Qu was placed in an incubator and the fermentation temperature was set to 37 °C, 42 °C and 50 °C, respectively. The culture was maintained for 7 d, and after fermentation was complete, the wheat Qu was fully dried for use.
2.5. Determination of Enzyme Activity in Cooked Wheat Qu
2.6. Transcriptome Sequencing and Analysis
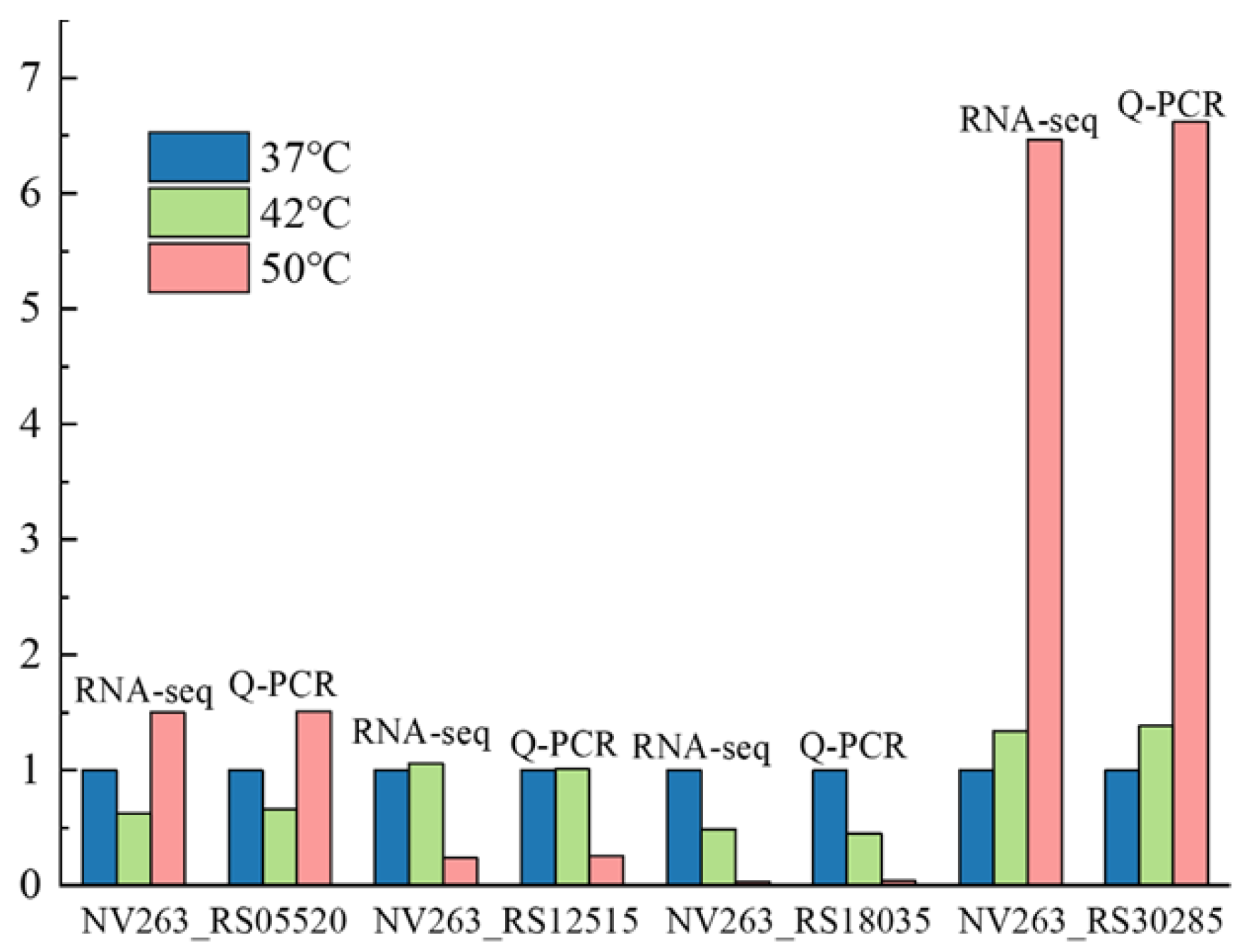
2.7. Primer Design and Gene Expression Verification
2.8. Statistical Analysis
3. Results and Discussion
3.1. Morphological Comparison of Saccharopolyspora rosea A22 at Different Temperatures
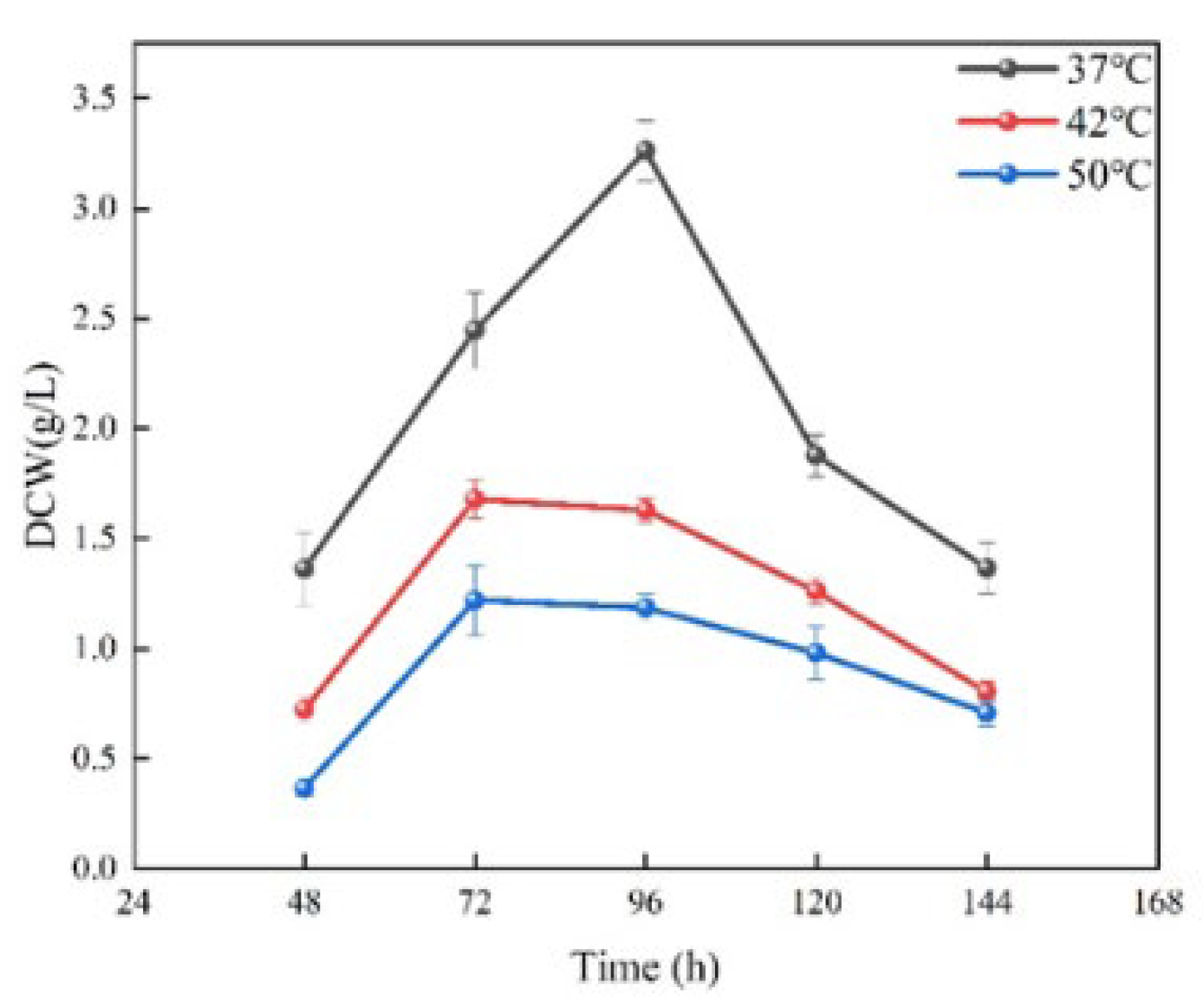
3.2. Biomass Differences in Saccharopolyspora rosea A22 at Different Temperatures
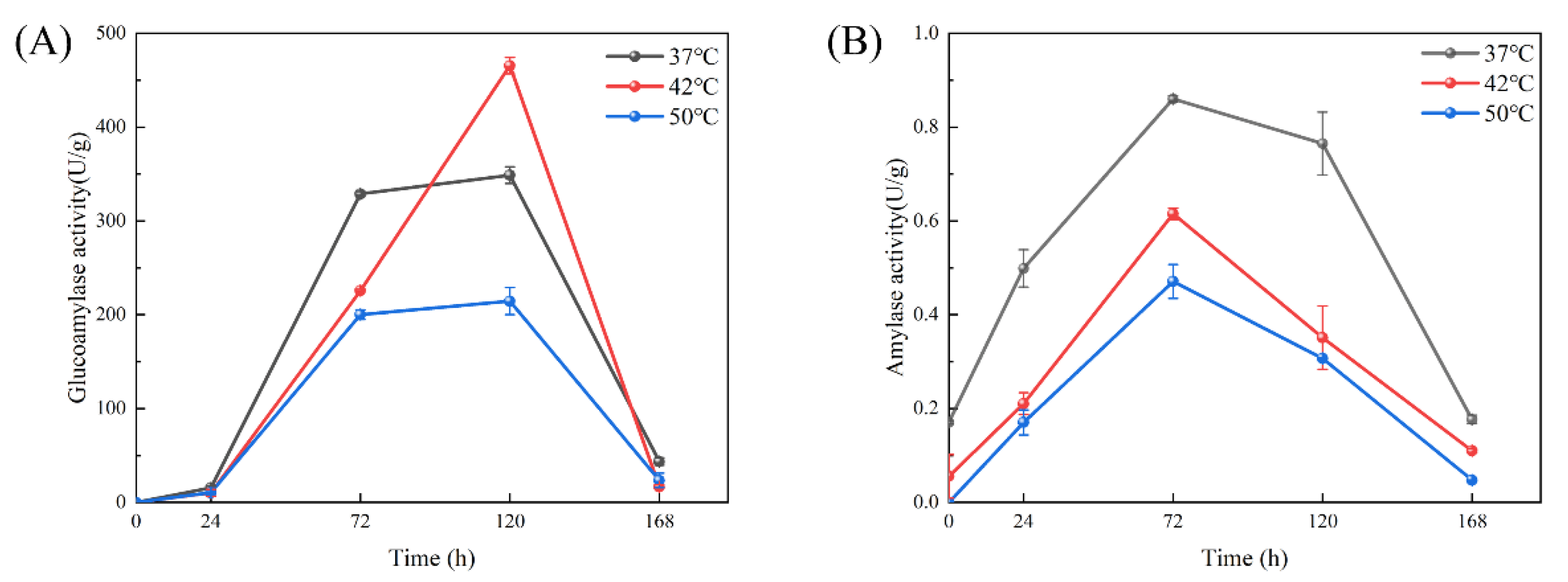
3.3. Comparison of Enzyme Activity in Preparation of Cooked Wheat Qu by Saccharopolyspora rosea A22 at Different Temperatures
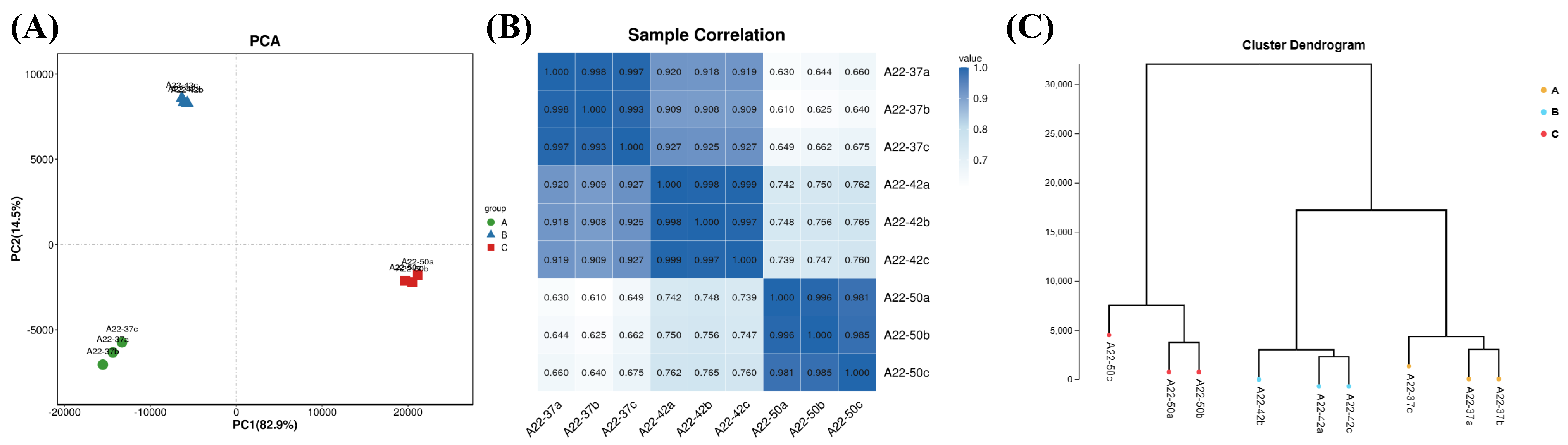
3.4. Transcriptome Sequencing Gene Statistics and Sample Relationship
3.5. Gene Expression Analysis
3.6. Functional Annotation of Differentially Expressed Genes
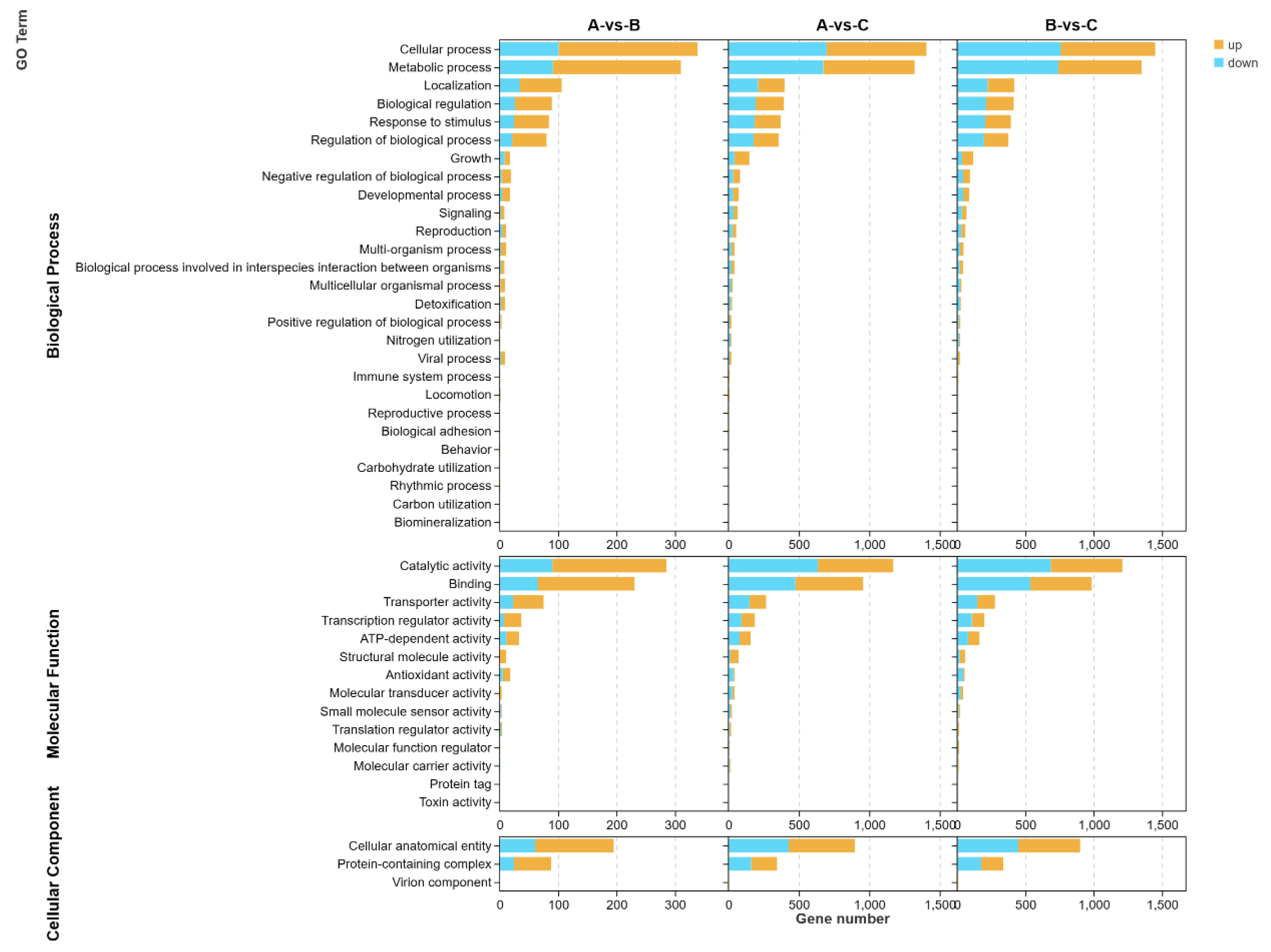
3.6.1. GO Analysis
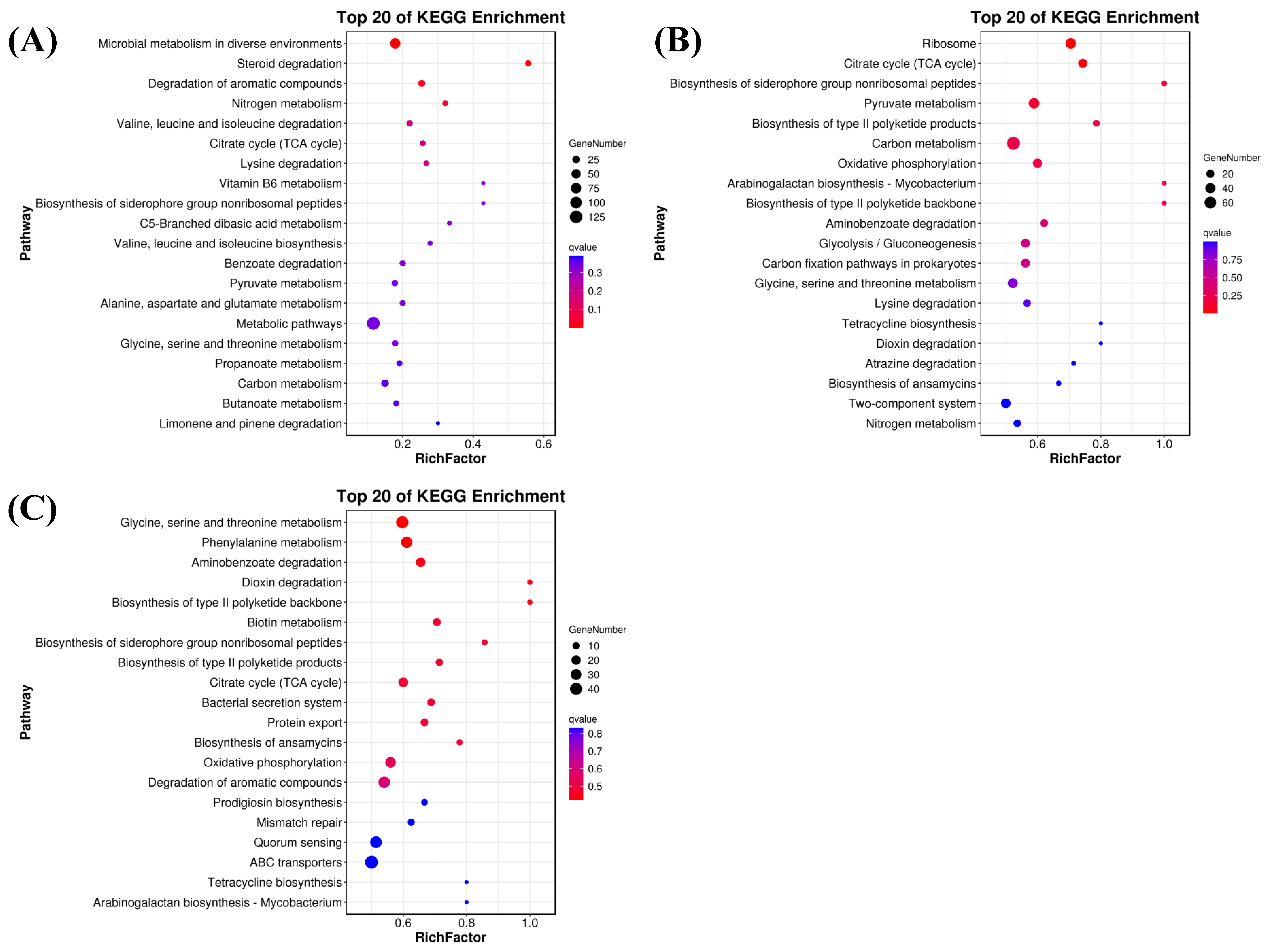
3.6.2. KEGG Analysis
3.7. Verification of Key Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, Z.R.; Guo, W.L.; Zhou, W.B.; Li, L.; Xu, J.X.; Hong, J.L.; Liu, H.P.; Zeng, F.; Bai, W.D.; Liu, B.; et al. Microbial Communities and Volatile Metabolites in Different Traditional Fermentation Starters Used for Hong Qu Glutinous Rice Wine. Food Res. Int. 2019, 121, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.F.; Zeng, W.Z.; Fang, F.; Zhou, J.W.; Du, G.C. The Microbiome of Chinese Rice Wine (Huangjiu). Curr. Res. Food Sci. 2022, 5, 325–335. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Y.; Qian, M.C. Comparison of the Aromatic Profile of Traditional and Modern Types of Huang Jiu (Chinese Rice Wine) by Aroma Extract Dilution Analysis and Chemical Analysis. Flavour Fragr. J. 2018, 33, 263–271. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Y. Effect of “Wheat Qu” on the Fermentation Processes and Volatile Flavour-Active Compounds of Chinese Rice Wine (Huangjiu). J. Inst. Brew. 2013, 119, 71–77. [Google Scholar] [CrossRef]
- Mo, X.L.; Xu, Y.; Fan, W. Characterization of Aroma Compounds in Chinese Rice Wine Qu by Solvent-Assisted Flavor Evaporation and Headspace Solid-Phase Microextraction. J. Agric. Food Chem. 2010, 58, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wang, L.; Zhang, Y.G.; Tu, T.Y.; Wang, S.T.; Shen, C.H.; Yuan, H.W.; Zhong, X.Z. A Comparison of Microbial Communities and Volatile Compounds in Wheat Qu from Different Geographic Locations. LWT-Food Sci. Technol. 2021, 148, 111752. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, S.P.; Sun, H.L.; Jiang, Z.F.; Xu, Y.Z.; Mao, J.Q.; Qian, B.; Wang, L.; Mao, J. Metagenomics-Based Insights into the Microbial Community Profiling and Flavor Development Potentiality of Baijiu Daqu and Huangjiu Wheat Qu. Food Res. Int. 2022, 152, 110707. [Google Scholar] [CrossRef]
- Häkkinen, M.; Valkonen, M.J.; Westerholm-Parvinen, A.; Aro, N.; Arvas, M.; Vitikainen, M.; Penttilä, M.; Saloheimo, M.; Pakula, T.M. Screening of Candidate Regulators for Cellulase and Hemicellulase Production in Trichoderma Reesei and Identification of a Factor Essential for Cellulase Production. Biotechnol. Biofuels 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.W.; Yu, X.; Wang, L.Y.; Zou, M.L.; Ma, J.Y.; Liu, J.; Feng, Y.L.; Zhao, S.M.; Yang, Q.; Hu, Y.L.; et al. Unveiling the Microbiota of Sauce-Flavor Daqu and Its Relationships with Flavors and Color during Maturation. Front. Microbiol. 2024, 15, 1345772. [Google Scholar] [CrossRef]
- do Nascimento, R.P.; Reis, A.D.; Gírio, F.; Pereira, N., Jr.; da Bon, E.P.S.; Coelho, R.R.R. A Thermotolerant Xylan-Degrading Enzyme Is Produced by Streptomyces Malaysiensis AMT-3 Using By-Products from the Food Industry. Braz. Arch. Biol. Technol. 2020, 63, e20190243. [Google Scholar] [CrossRef]
- Wang, Y.; Quan, S.K.; Zhao, Y.J.; Xia, Y.; Zhang, R.; Ran, M.F.; Wu, Z.Y.; Zhang, W.X. The Active Synergetic Microbiota with Aspergillus as the Core Dominates the Metabolic Network of Ester Synthesis in Medium-High Temperature Daqu. Food Microbiol. 2023, 115, 104336. [Google Scholar] [CrossRef] [PubMed]
- El-Tarabily, K.A.; Soliman, M.H.; Nassar, A.H.; Al-Hassani, H.A.; Sivasithamparam, K.; McKenna, F.; Hardy, G.E.J. Biological Control of Sclerotinia Minor Using a Chitinolytic Bacterium and Actinomycetes. Plant Pathol. 2000, 49, 573–583. [Google Scholar] [CrossRef]
- Wang, B.W.; Wu, Q.; Xu, Y.; Sun, B.G. Specific Volumetric Weight-Driven Shift in Microbiota Compositions with Saccharifying Activity Change in Starter for Chinese Baijiu Fermentation. Front. Microbiol. 2018, 9, 2349. [Google Scholar] [CrossRef] [PubMed]
- Zeeman, S.C.; Kossmann, J.; Smith, A.M. Starch: Its Metabolism, Evolution, and Biotechnological Modification in Plants. Annu. Rev. Plant Biol. 2010, 61, 209–234. [Google Scholar] [CrossRef]
- Lacey, J.; Goodfellow, M. A Novel Actinomycete from Sugar-Cane Bagasse: Saccharopolyspora hirsuta Gen. Et Sp. Nov. J. Gen. Microbiol. 1975, 88, 75–85. [Google Scholar] [CrossRef]
- Sayed, A.M.; Abdel-Wahab, N.M.; Hassan, H.M.; Abdelmohsen, U.R. Saccharopolyspora: An Underexplored Source for Bioactive Natural Products. J. Appl. Microbiol. 2019, 128, 314–329. [Google Scholar] [CrossRef]
- Huang, Z.G.; Zeng, B.; Deng, J.; Ren, Z.Q.; Xie, J.; Wei, C.H. Succession of Microbial Community Structure in Fermented Grains during the Fermentation of Strong-Flavor Baijiu and Its Impact on the Metabolism of Acids, Alcohols, and Esters. Food Sci. Biotechnol. 2024. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Y.G.; Huang, Y.L. Microbiome Diversity and Evolution in Stacking Fermentation during Different Rounds of Jiang-Flavoured Baijiu Brewing. LWT 2021, 143, 111119. [Google Scholar] [CrossRef]
- Liu, M.K.; Tang, Y.M.; Zhao, K.; Liu, Y.; Guo, X.J.; Tian, X.H.; Ren, D.Q.; Yao, W.C. Contrasting Bacterial Community Structure in Artificial Pit Mud-Starter Cultures of Different Qualities: A Complex Biological Mixture for Chinese Strong-Flavor Baijiu Production. 3 Biotech 2019, 9, 89. [Google Scholar] [CrossRef]
- Xu, S.S.; Zhang, M.Z.; Xu, B.Y.; Liu, L.H.; Sun, W.; Mu, D.D.; Wu, X.F.; Li, X.J. Microbial Communities and Flavor Formation in the Fermentation of Chinese Strong-Flavor Baijiu Produced from Old and New Zaopei. Food Res. Int. 2022, 156, 111162. [Google Scholar] [CrossRef]
- Deng, L.; Mao, X.; Liu, D.; Ning, X.Q.; Shen, Y.; Chen, B.; Nie, H.F.; Huang, D.; Luo, H.B. Comparative Analysis of Physicochemical Properties and Microbial Composition in High-Temperature Daqu with Different Colors. Front. Microbiol. 2020, 11, 588117. [Google Scholar] [CrossRef]
- Shang, C.H.; Li, Y.J.; Zhang, J.; Gan, S.L. Analysis of Bacterial Diversity in Different Types of Daqu and Fermented Grains from Danquan Distillery. Front. Microbiol. 2022, 13, 883122. [Google Scholar] [CrossRef]
- Wang, P.X.; Mao, J.; Meng, X.Y.; Li, X.Z.; Liu, Y.Y.; Feng, H. Changes in Flavour Characteristics and Bacterial Diversity during the Traditional Fermentation of Chinese Rice Wines from Shaoxing Region. Food Control 2014, 44, 58–63. [Google Scholar] [CrossRef]
- Guan, Z.B.; Zhang, Z.H.; Cao, Y.; Chen, L.L.; Xie, G.F.; Lu, J. Analysis and Comparison of Bacterial Communities in Two Types of “WheatQu”, the Starter Culture of Shaoxing Rice Wine, Using Nested PCR-DGGE. J. Inst. Brew. 2012, 118, 127–132. [Google Scholar] [CrossRef]
- Liu, S.P.; Mao, J.; Liu, Y.Y.; Meng, X.Y.; Ji, Z.W.; Zhou, Z.L.; Ai-lati, A. Bacterial Succession and the Dynamics of Volatile Compounds during the Fermentation of Chinese Rice Wine from Shaoxing Region. World J. Microbiol. Biotechnol. 2015, 31, 1907–1921. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.T.; Chen, J.; Liu, L.; Wu, H.; Tan, H.Q.; Xie, G.F.; Xu, Q.; Zou, H.J.; Yu, W.J.; Wang, L.; et al. Metagenomic Sequencing Reveals the Relationship between Microbiota Composition and Quality of Chinese Rice Wine. Sci. Rep. 2016, 6, 26621. [Google Scholar] [CrossRef]
- Gan, S.H.; Yang, F.; Sahu, S.K.; Luo, R.; Liao, S.L.; Wang, H.Y.; Jin, T.; Wang, L.; Zhang, P.F.; Liu, X.; et al. Deciphering the Composition and Functional Profile of the Microbial Communities in Chinese Moutai Liquor Starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.D.; Xie, G.F.; Wu, D.H.; Yang, L.X.; Jin, Z.; Hu, Z.M.; Xu, X.B.; Lu, J. Isolation and Identification of the Bitter Compound from Huangjiu. Food Chem. 2021, 349, 129133. [Google Scholar] [CrossRef]
- Ma, D.L.; Liu, S.P.; Liu, H.P.; Nan, M.J.; Xu, Y.Z.; Han, X.; Mao, J. Developing an Innovative Raw Wheat Qu Inoculated with Saccharopolyspora and Its Application in Huangjiu. J. Sci. Food Agric. 2022, 102, 7301–7312. [Google Scholar] [CrossRef]
- Liu, S.P.; Chen, Q.L.; Zou, H.J.; Yu, Y.J.; Zhou, Z.L.; Mao, J.; Zhang, S. A Metagenomic Analysis of the Relationship between Microorganisms and Flavor Development in Shaoxing Mechanized Huangjiu Fermentation Mashes. Int. J. Food Microbiol. 2019, 303, 9–18. [Google Scholar] [CrossRef]
- Zhu, M.; Zheng, J.; Xie, J.; Zhao, D.; Qiao, Z.W.; Huang, D.; Luo, H.B. Effects of Environmental Factors on the Microbial Community Changes during Medium-High Temperature Daqu Manufacturing. Food Res. Int. 2022, 153, 110955. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Luo, H.B.; Zhao, D.; Qiao, Z.W.; Zheng, J.; An, M.Z.; Huang, D. Environmental Factors and Interactions among Microorganisms Drive Microbial Community Succession during Fermentation of Nongxiangxing Daqu. Bioresour. Technol. 2022, 345, 126549. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, A.; Hames, B.; Hyman, D.; Payne, C.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Wolfe, J. Determination of Total Solids in Biomass and Total Dissolved Solids in Liquid Process Samples; NREL Technical Report No. NREL/TP-510-42621; National Renewable Energy Laboratory: Golden, CO, USA, 2008. [Google Scholar]
- Fu, J. Analysis of Wheat Qu and Hongqu (Wuyihongqu). In Huangjiu Brewing Technology; Chemical Industry Press: Beijing, China, 2005; pp. 412–416. [Google Scholar]
- Liu, S.P.; Hu, J.; Xu, Y.Z.; Xue, J.B.; Zhou, J.D.; Han, X.; Ji, Z.W.; Mao, J. Combined Use of Single Molecule Real-Time DNA Sequencing Technology and Culture-Dependent Methods to Analyze the Functional Microorganisms in Inoculated Raw Wheat Qu. Food Res. Int. 2020, 132, 109062. [Google Scholar] [CrossRef]
- Wang, P.; Li, B.Q.; Li, B.Y.; Yang, J.; Xu, X.R.; Yang, S.T.; Zou, X. Carbon-Economic Biosynthesis of Poly-2-Hydrobutanedioic Acid Driven by Nonfermentable Substrate Ethanol. Green Chem. 2022, 24, 6599–6612. [Google Scholar] [CrossRef]
- Peng, Q.; Zheng, H.J.; Yu, H.F.; Meng, K.; Cheng, Y.J.; Yang, X.Y.; Xie, G.F.; Zheng, X.M. Environmental Factors Drive the Succession of Microbial Community Structure during Wheat Qu Fermentation. Food Biosci. 2023, 56, 103169. [Google Scholar] [CrossRef]
- Du, H.; Lu, H.; Xu, Y.; Du, X.W. Community of Environmental Streptomyces Related to Geosmin Development in Chinese Liquors. J. Agric. Food Chem. 2013, 61, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, W.; Yang, Q. Transcriptome Analysis Reveals the Regulation of Aureobasidium Pullulans under Different PH Stress. Int. J. Mol. Sci. 2023, 24, 16103. [Google Scholar] [CrossRef]
- Jung, J.Y.; Lee, S.H.; Kim, J.M.; Park, M.S.; Bae, J.-W.; Hahn, Y.; Madsen, E.L.; Jeon, C.O. Metagenomic Analysis of Kimchi, a Traditional Korean Fermented Food. Appl. Environ. Microbiol. 2011, 77, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese Rind Communities Provide Tractable Systems for in Situ and in Vitro Studies of Microbial Diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takumi, K.; Miki, K. Crystal Structure of a Thermophilic GrpE Protein: Insight into Thermosensing Function for the DnaK Chaperone System. J. Mol. Biol. 2010, 396, 1000–1011. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Name | Sequence (5′ to 3′) |
|---|---|---|
| Amy1F(NV263_RS18035) | Amy1F | CGCTGACCTGAAAACCGAA |
| Amy1R | TGACCGTTGAAGGTGTAGC | |
| Amy2F(NV263_RS12515) | Amy2F | ATGGCTTCCTCGCTGGTCA |
| Amy2R | GTTGCTGCCCTCCTTGTCC | |
| ClpXF(NV263_RS05520) | ClpXF | GATGAGCCCGTACTTGATC |
| ClpXR | CGGCTGCGGCAAGACCTAC | |
| DnaB(NV263_RS30285) | DnaBF | GCTACCACGGCTCCGAGGG |
| DnaBR | CGGGAGAACTCGGAGACTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.; Lu, P.; Ma, J.; Li, Z.; Han, X.; Ji, Z.; Liu, S.; Mao, J. Transcriptome Analysis Reveals the Variations in Enzyme Production of Saccharopolyspora rosea A22 under Different Temperatures. Foods 2024, 13, 2696. https://doi.org/10.3390/foods13172696
Lin C, Lu P, Ma J, Li Z, Han X, Ji Z, Liu S, Mao J. Transcriptome Analysis Reveals the Variations in Enzyme Production of Saccharopolyspora rosea A22 under Different Temperatures. Foods. 2024; 13(17):2696. https://doi.org/10.3390/foods13172696
Chicago/Turabian StyleLin, Congyu, Peiqi Lu, Jingqiu Ma, Zhihui Li, Xiao Han, Zhongwei Ji, Shuangping Liu, and Jian Mao. 2024. "Transcriptome Analysis Reveals the Variations in Enzyme Production of Saccharopolyspora rosea A22 under Different Temperatures" Foods 13, no. 17: 2696. https://doi.org/10.3390/foods13172696
APA StyleLin, C., Lu, P., Ma, J., Li, Z., Han, X., Ji, Z., Liu, S., & Mao, J. (2024). Transcriptome Analysis Reveals the Variations in Enzyme Production of Saccharopolyspora rosea A22 under Different Temperatures. Foods, 13(17), 2696. https://doi.org/10.3390/foods13172696