Illuminating the Characteristics and Assembly of Prokaryotic Communities across a pH Gradient in Pit Muds for the Production of Chinese Strong-Flavor Baijiu

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Analysis of Physicochemical Properties
2.2. DNA Extraction, Amplification, Illumina Sequencing, and Sequence Processing
2.3. Statistical Analysis
2.4. Microbial Network Construction and Characterization
2.5. The Phylogenetic-Bin-Based Null Model Analysis
3. Results
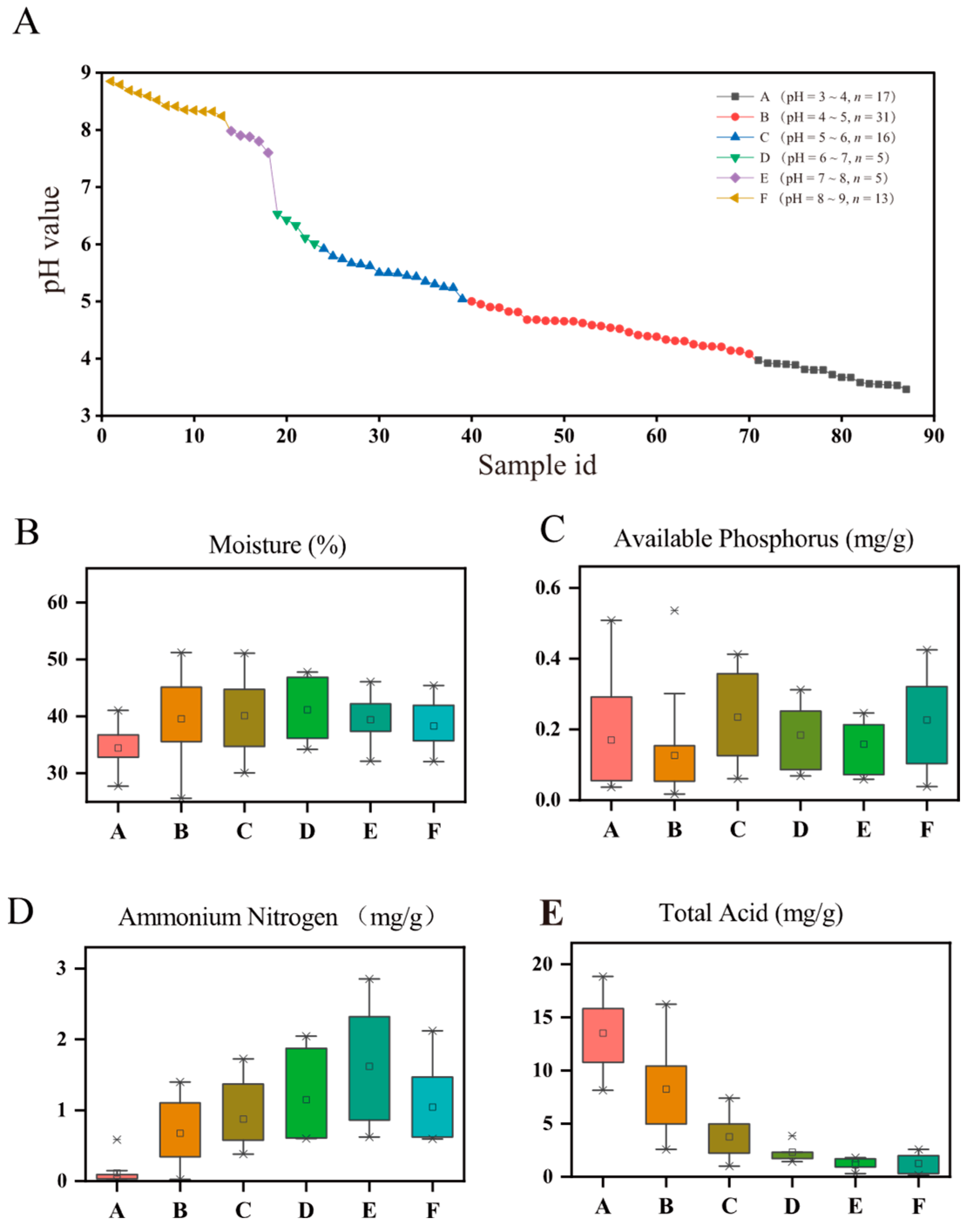
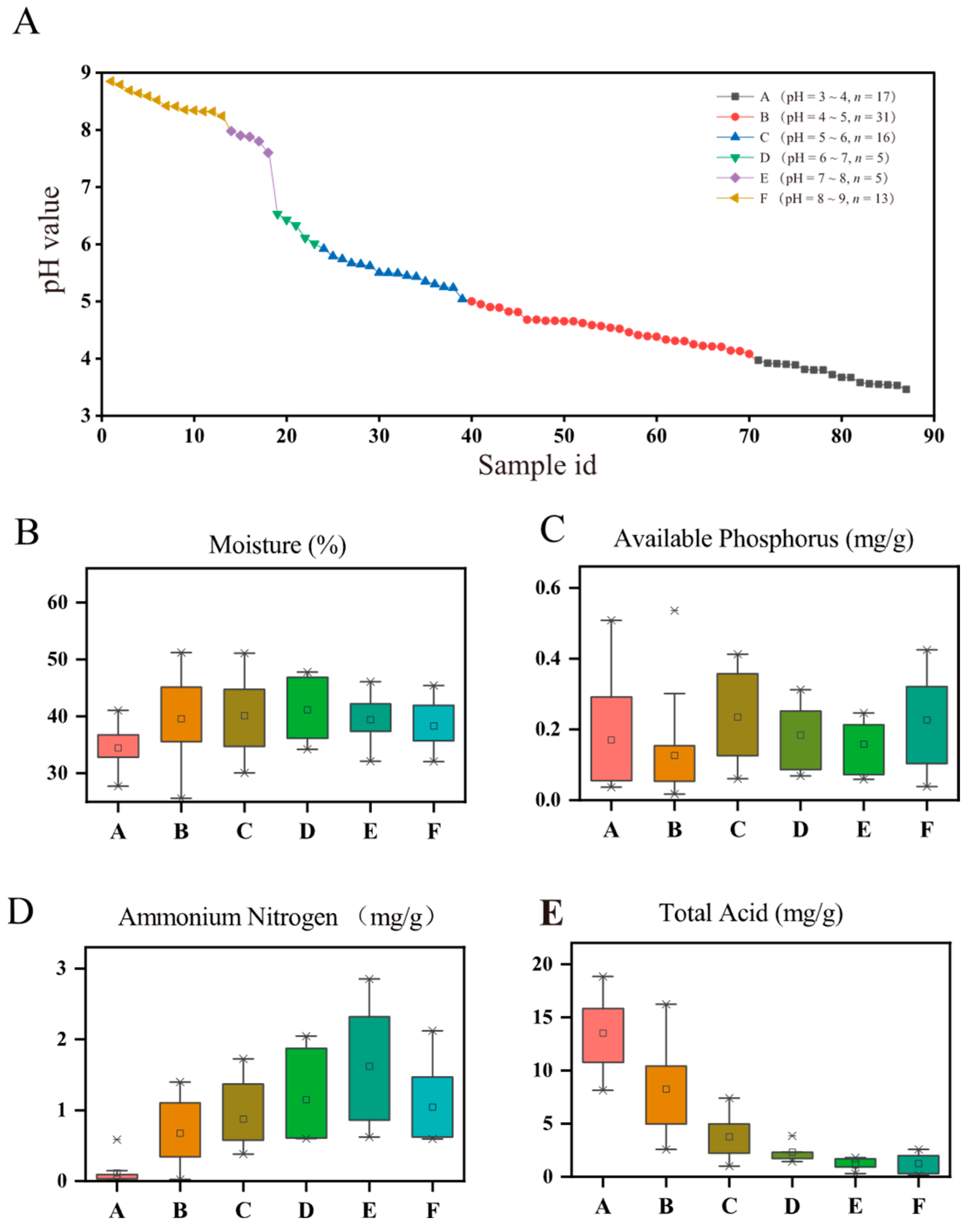
3.1. Physicochemical Properties in PMs across a pH Gradient
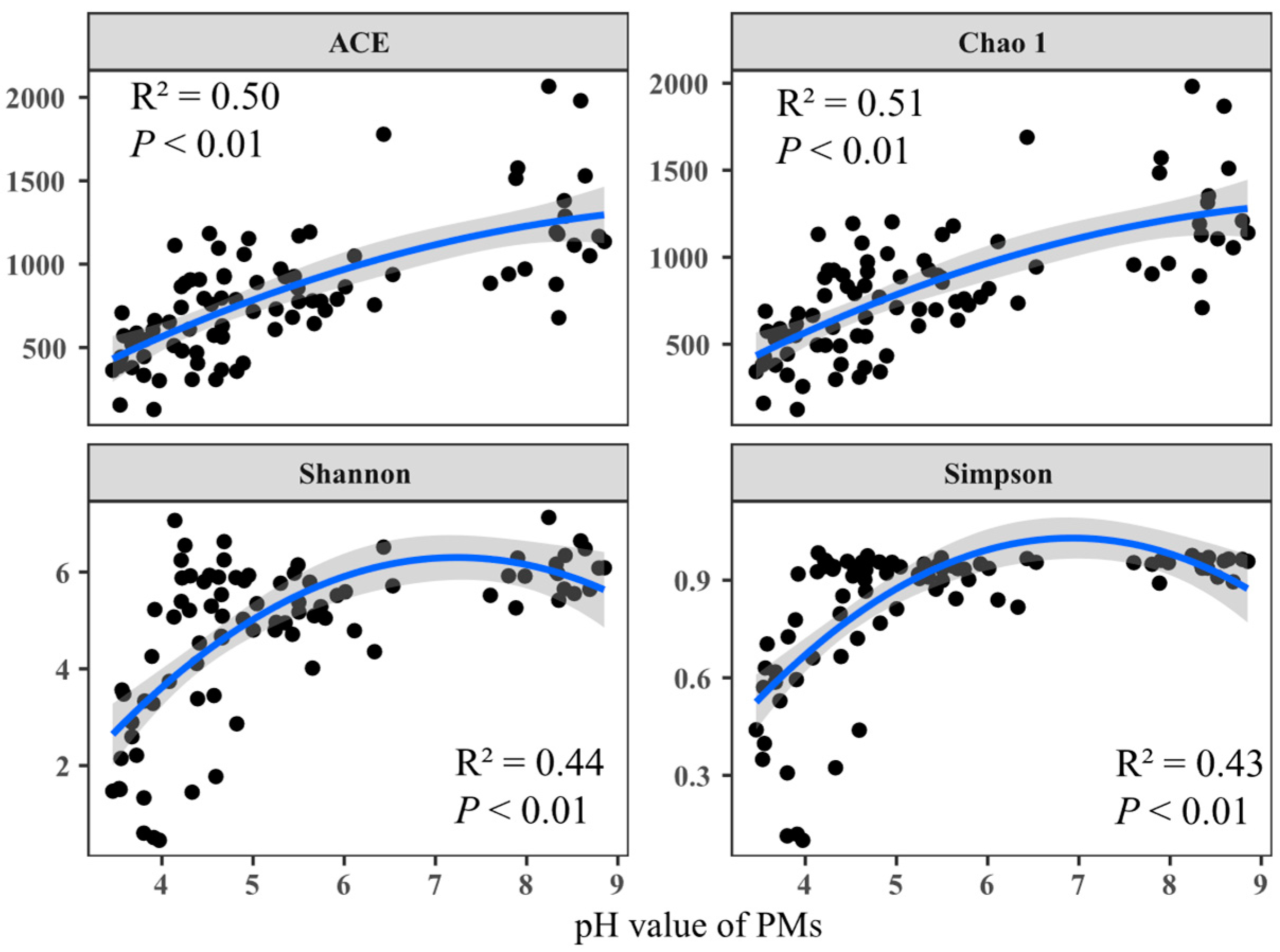
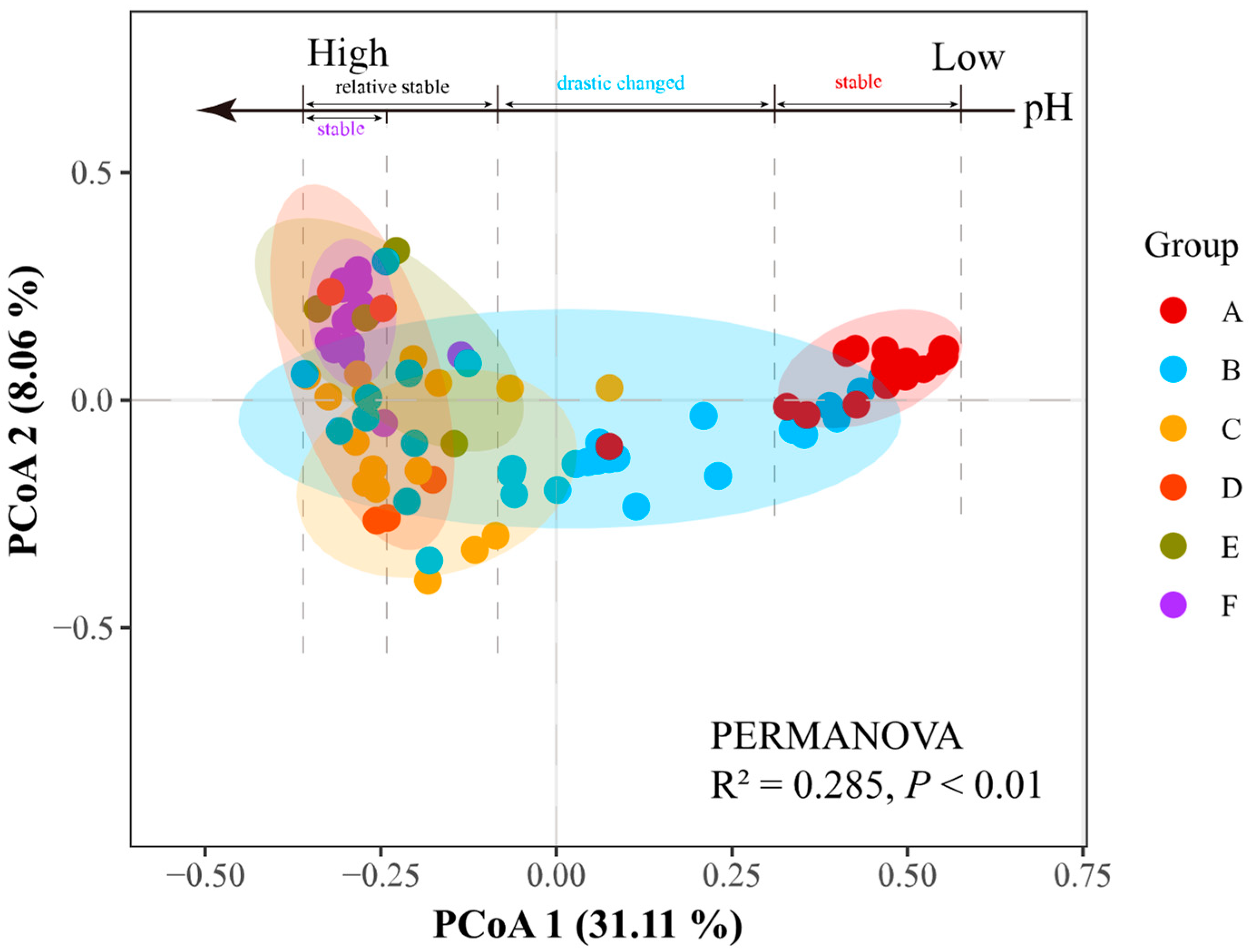
3.2. Variation in α- and β-Diversities of Prokaryotic Communities in PMs across a pH Gradient
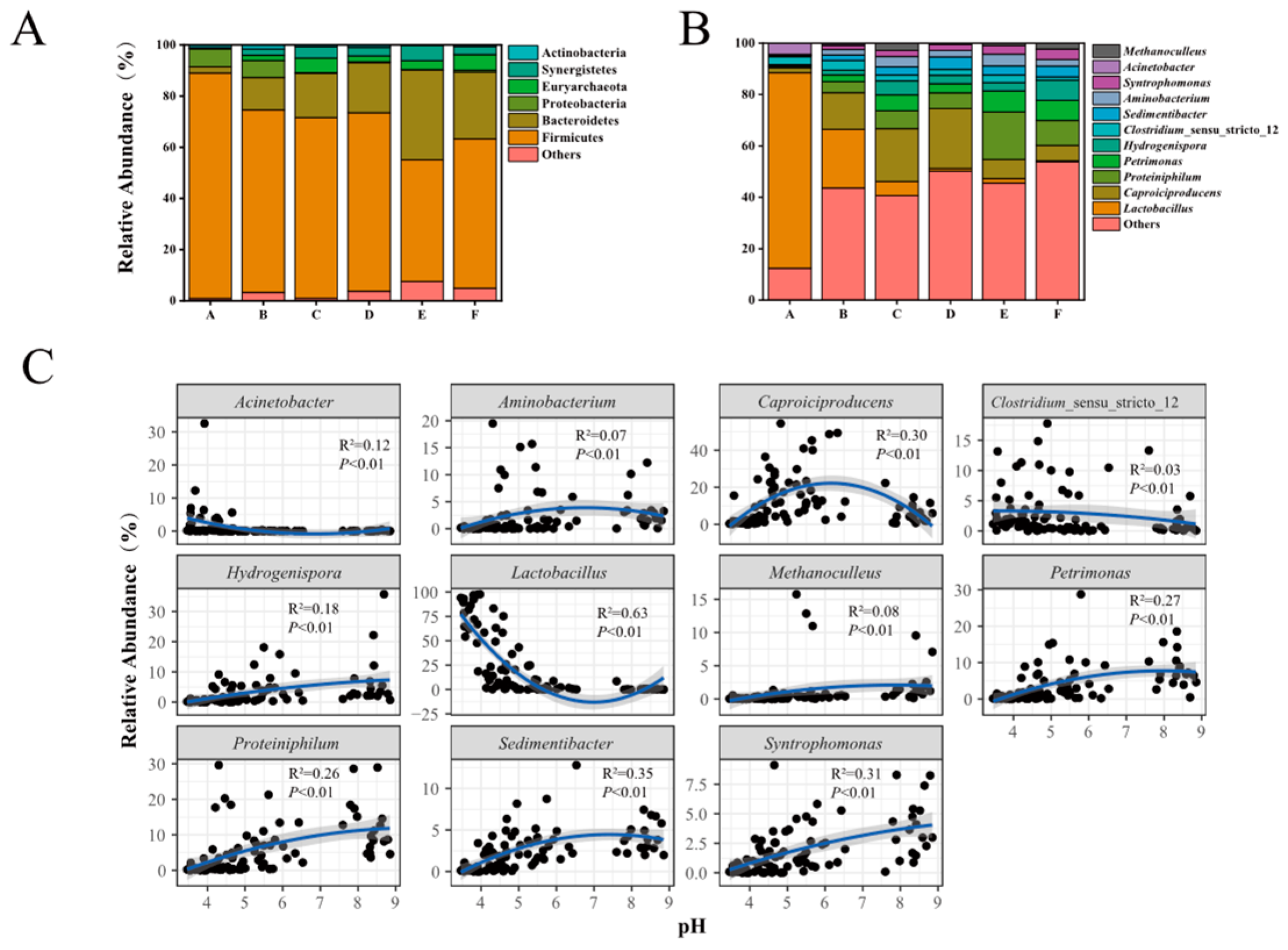
3.3. The Overall Features of Prokaryotic Community Structure in PMs
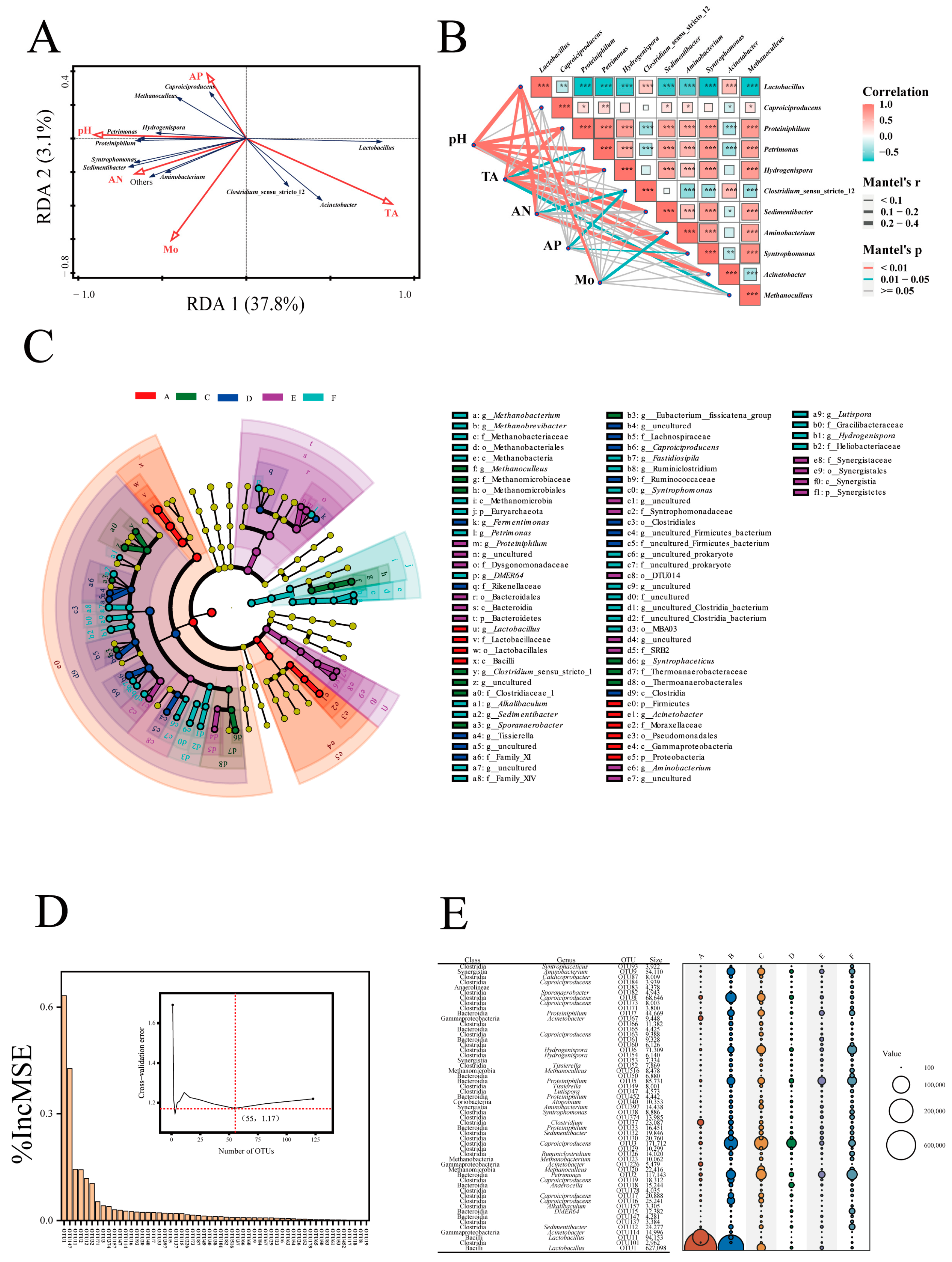
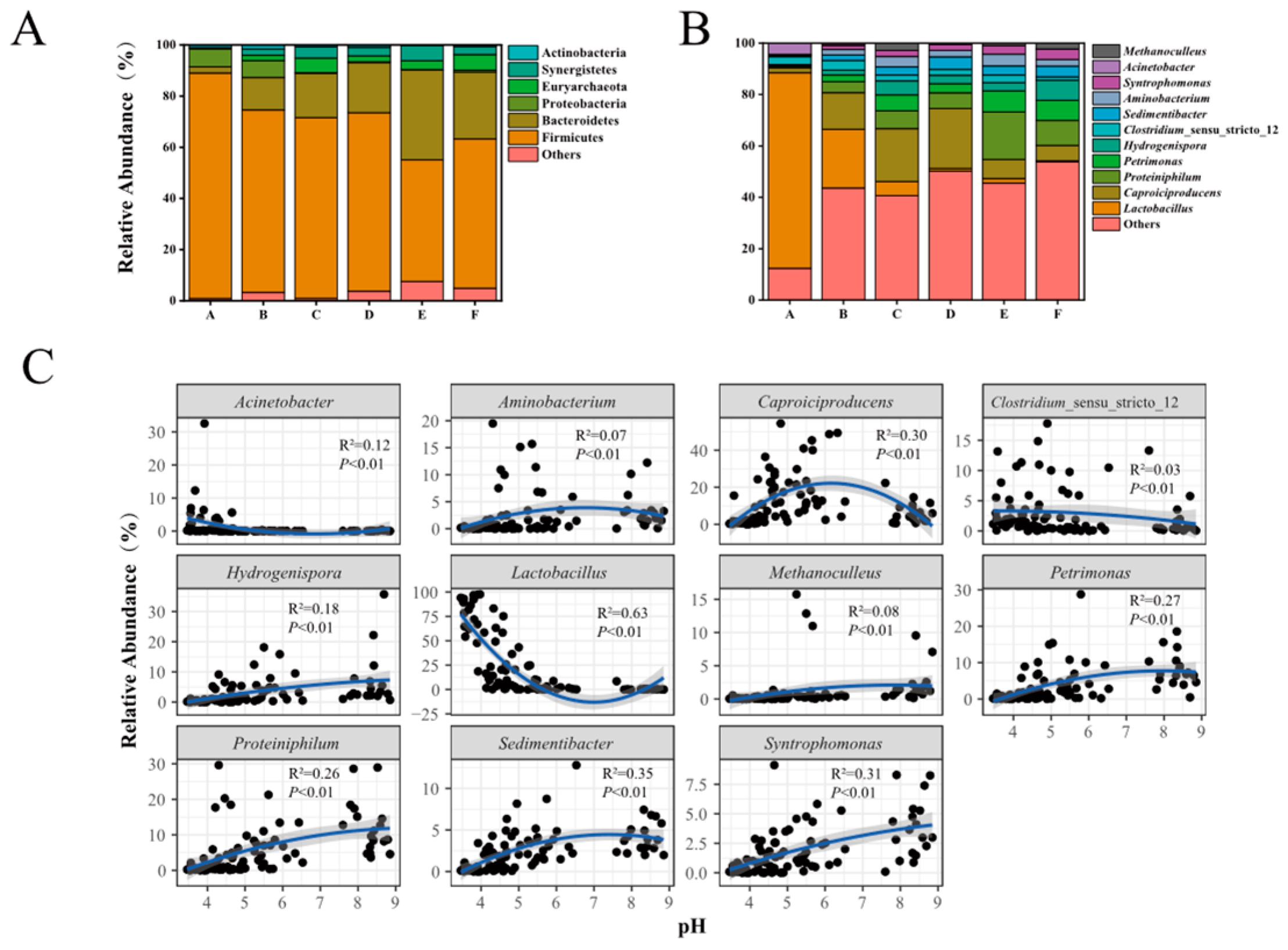
3.4. Effects of Physicochemical Factors on Prokaryotic Community and Biomarkers in PMs
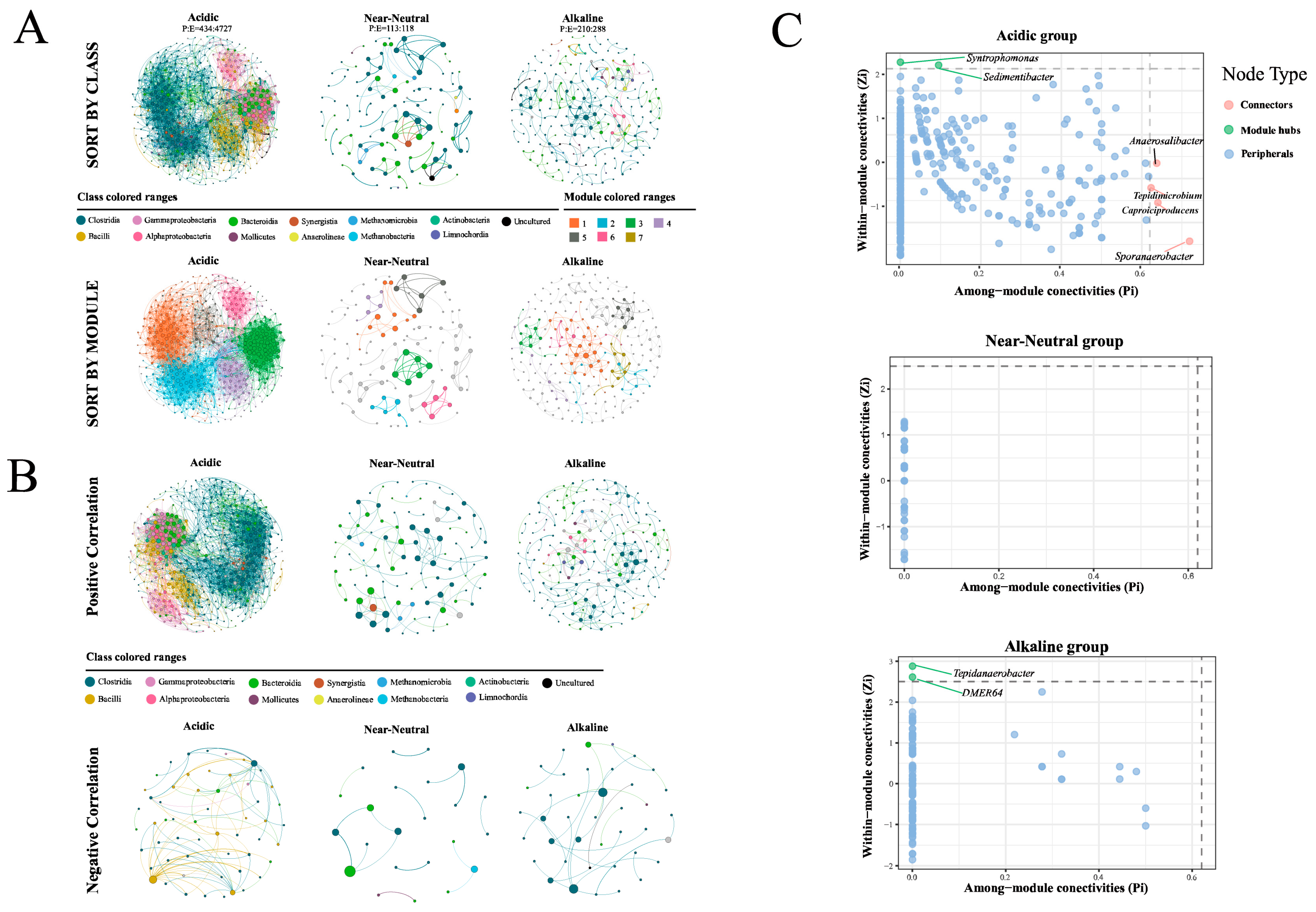
3.5. The Network Patterns of Prokaryotic Communities in PMs with Different pHs
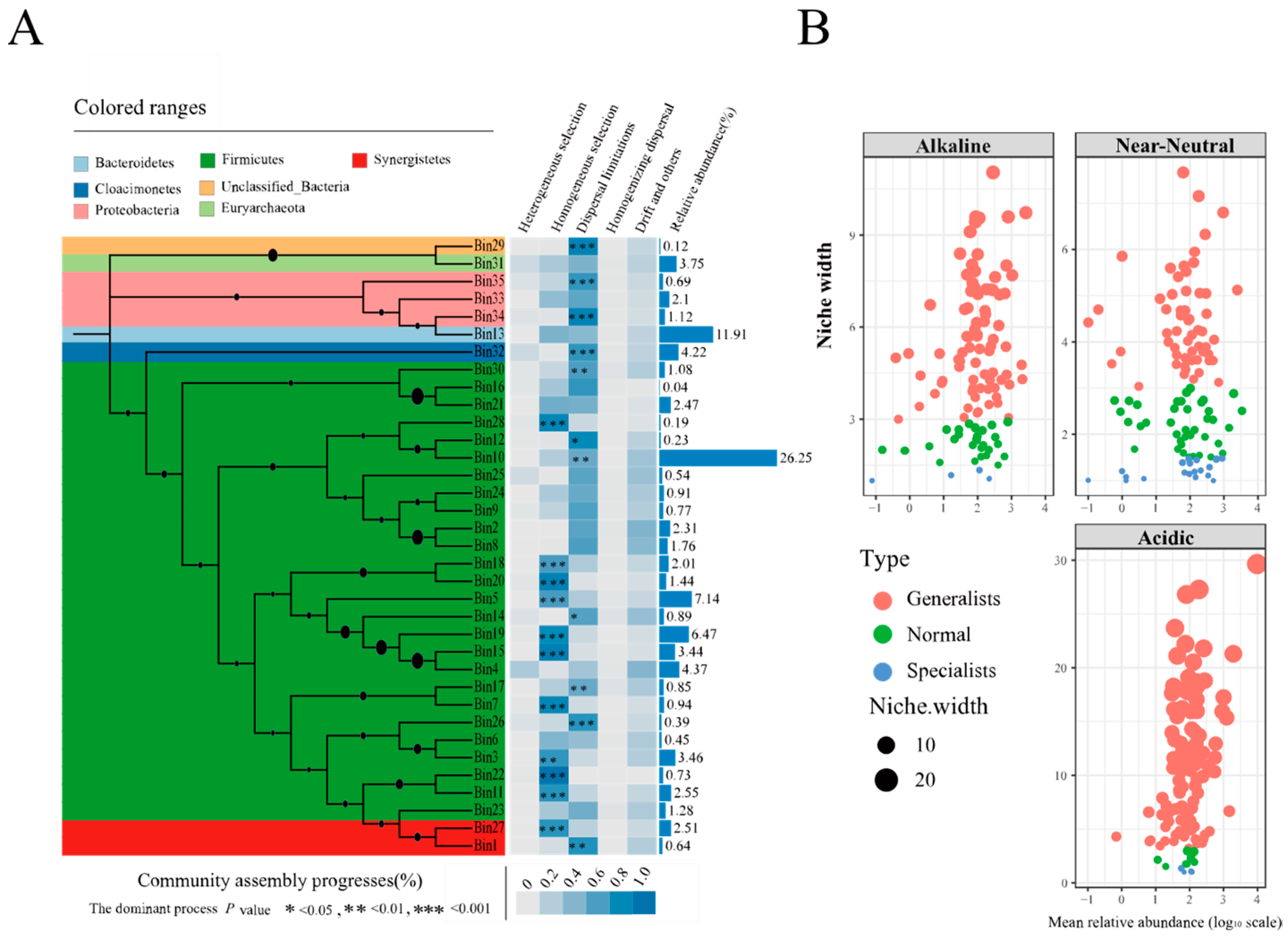
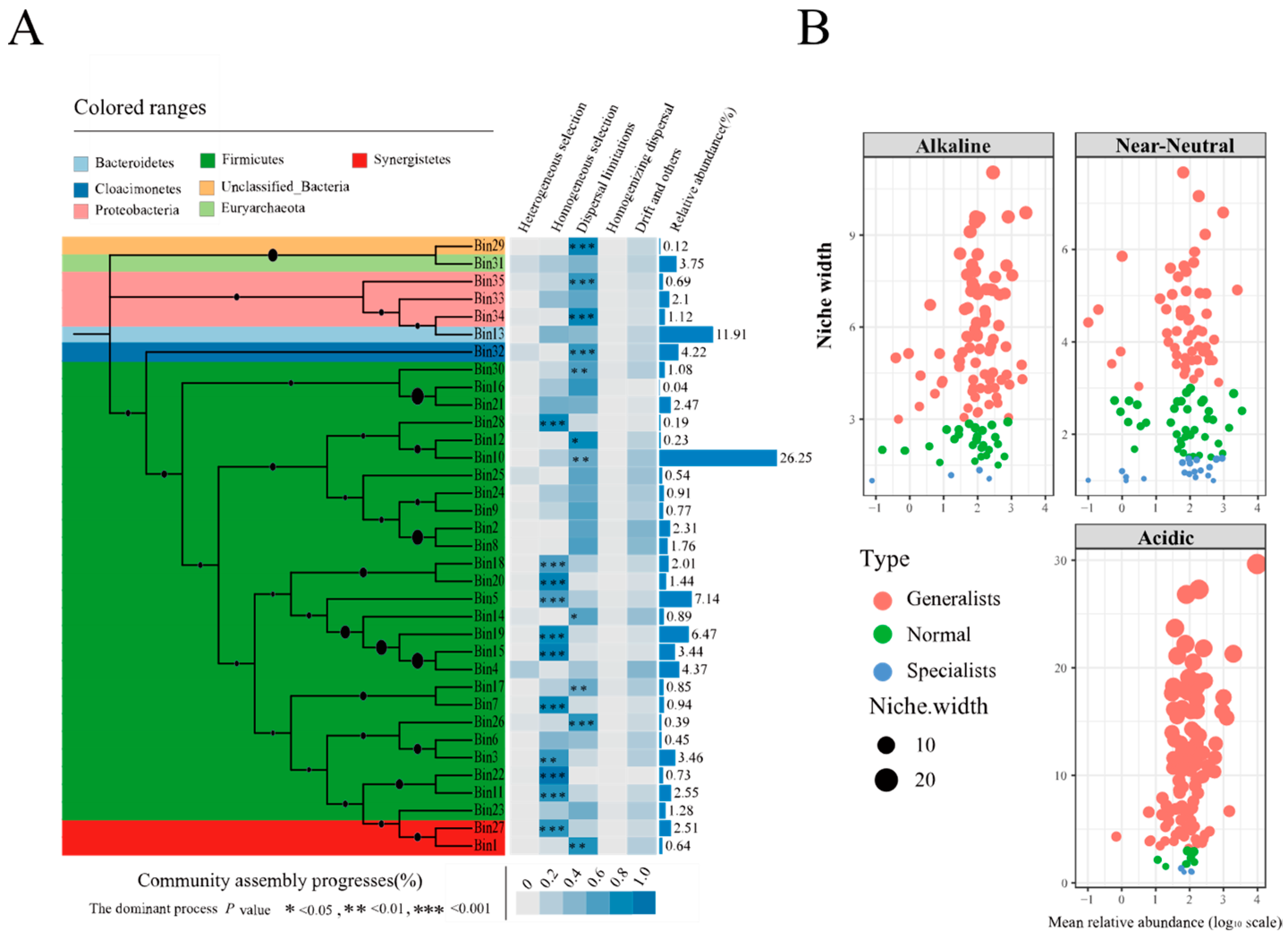
3.6. The Assembly Process of Prokaryotic Communities in PMs
4. Discussion
4.1. pH as a Predictor of α- and β-Diversities of Prokaryotic Communities in PMs
4.2. Variation Characteristics of Prokaryotic Community Structures and Biomarker Identification across a pH Gradient in PMs
4.3. Effect of pH on the Network Characteristics of Prokaryotic Communities in PMs
4.4. The Assembly Mechanisms of Prokaryotic Communities in PMs Based on iCAMP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGovern, P.E.; Zhang, J.; Tang, J.; Zhang, Z.; Hall, G.R.; Moreau, R.A.; Nuñez, A.; Butrym, E.D.; Richards, M.P.; Wang, C.S.; et al. Fermented beverages of pre- and proto-historic China. Proc. Natl. Acad. Sci. USA 2004, 101, 17593–17598. [Google Scholar] [CrossRef] [PubMed]
- Chilton, S.N.; Burton, J.P.; Reid, G. Inclusion of fermented foods in food guides around the world. Nutrients 2015, 7, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.L.; Chai, L.J.; Zhong, X.Z.; Lu, Z.M.; Zhang, X.J.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Microbial biogeography of pit mud from an artificial brewing ecosystem on a large time scale: All roads lead to Rome. mSystems 2023, 8, e0056423. [Google Scholar] [CrossRef]
- Tao, Y.; Li, J.; Rui, J.; Xu, Z.; Zhou, Y.; Hu, X.; Wang, X.; Liu, M.; Li, D.; Li, X. Prokaryotic communities in pit mud from different-aged cellars used for the production of Chinese strong-flavored liquor. Appl. Environ. Microbiol. 2014, 80, 2254–2260. [Google Scholar] [CrossRef]
- Ren, C.; Gu, Y.; Du, H.; Xu, Y. Predicting dominant caproate-producing microbes by comparing the microbiotas between new-and aged-pit muds. Food Ferment. Ind. 2018, 44, 8–14. [Google Scholar] [CrossRef]
- Thauer, R.K.; Jungermann, K.; Henninger, H.; Wenning, J.; Decker, K. The energy metabolism of Clostridium kluyveri. Eur. J. Biochem. 1968, 4, 173–180. [Google Scholar] [CrossRef]
- Chai, L.J.; Qian, W.; Zhong, X.Z.; Zhang, X.J.; Lu, Z.M.; Zhang, S.Y.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Mining the Factors Driving the Evolution of the Pit Mud Microbiome under the Impact of Long-Term Production of Strong-Flavor Baijiu. Appl. Environ. Microbiol. 2021, 87, e00885-21. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.Y.; Hou, C.J.; Bian, M.H.; Shen, C.H.; Zhang, S.Y.; Huo, D.Q.; Ma, Y. Characterization of microbial community profiles associated with quality of Chinese strong-aromatic liquor through metagenomics. J. Appl. Microbiol. 2019, 127, 750–762. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, Y.; Wang, Y.; Wu, T.; Li, X.; Li, D.; Tao, Y. Production of high-concentration n-caproic acid from lactate through fermentation using a newly isolated Ruminococcaceae bacterium CPB6. Biotechnol. Biofuels 2017, 10, 102. [Google Scholar] [CrossRef]
- Wang, H.; Gu, Y.; Zhou, W.; Zhao, D.; Qiao, Z.; Zheng, J.; Gao, J.; Chen, X.; Ren, C.; Xu, Y. Adaptability of a Caproate-Producing Bacterium Contributes to Its Dominance in an Anaerobic Fermentation System. Appl. Environ. Microbiol. 2021, 87, e0120321. [Google Scholar] [CrossRef]
- Zou, W.; Zhao, C.; Luo, H. Diversity and Function of Microbial Community in Chinese Strong-Flavor Baijiu Ecosystem: A Review. Front. Microbiol. 2018, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Meng, Y.; Wang, Y.; Zhou, Q.; Li, A.; Liu, G.; Li, J.; Xing, X. Prokaryotic communities in multidimensional bottom-pit-mud from old and young pits used for the production of Chinese Strong-Flavor Baijiu. Food Chem. 2020, 312, 126084. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.K.; Tang, Y.M.; Guo, X.J.; Zhao, K.; Penttinen, P.; Tian, X.H.; Zhang, X.Y.; Ren, D.Q.; Zhang, X.P. Structural and Functional Changes in Prokaryotic Communities in Artificial Pit Mud during Chinese Baijiu Production. mSystems 2020, 5, e00829-19. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yu, M.; Tian, R. Spatial Heterogeneity of Prokaryotic Microbial Community Diversity in Pit Mud from Degraded Cellar for the Production of Strong-flavor Baijiu Revealed by High Throughput Sequencing. Food Sci. 2021, 2021, 86–93. [Google Scholar] [CrossRef]
- Hu, X.; Du, H.; Ren, C.; Xu, Y. Illuminating Anaerobic Microbial Community and Cooccurrence Patterns across a Quality Gradient in Chinese Liquor Fermentation Pit Muds. Appl. Environ. Microbiol. 2016, 82, 2506–2515. [Google Scholar] [CrossRef]
- Liang, H.; Luo, Q.; Zhang, A.; Wu, Z.; Zhang, W. Comparison of bacterial community in matured and degenerated pit mud from ChineseLuzhou-flavourliquor distillery in different regions. J. Inst. Brew. 2016, 122, 48–54. [Google Scholar] [CrossRef]
- Liu, M.K.; Tang, Y.M.; Zhao, K.; Liu, Y.; Guo, X.J.; Tian, X.H.; Ren, D.Q.; Yao, W.C. Contrasting bacterial community structure in artificial pit mud-starter cultures of different qualities: A complex biological mixture for Chinese strong-flavor Baijiu production. 3 Biotech. 2019, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Hu, X. Illuminating the Correlation between Anaerobic Clostridial Community Diversity and Quality of Pit Mud Used for the Production of Chinese Strong-Flavor Liquor. Ph.D. Thesis, Jiangnan University, Wuxi City, China, 2015. [Google Scholar]
- Aciego Pietri, J.C.; Brookes, P.C. Relationships between soil pH and microbial properties in a UK arable soil. Soil Biol. Biochem. 2008, 40, 1856–1861. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef]
- Leibold, M.A.; Holyoak, M.; Mouquet, N.; Amarasekare, P.; Chase, J.M.; Hoopes, M.F.; Holt, R.D.; Shurin, J.B.; Law, R.; Tilman, D.; et al. The metacommunity concept: A framework for multi-scale community ecology. Ecol. Lett. 2004, 7, 601–613. [Google Scholar] [CrossRef]
- Lindström, E.S.; Langenheder, S. Local and regional factors influencing bacterial community assembly. Environ. Microbiol. Rep. 2012, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chave, J. Neutral theory and community ecology. Ecol. Lett. 2004, 7, 241–253. [Google Scholar] [CrossRef]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 2020, 11, 4717. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Huo, Q.B.; Wen, T.; Wang, X.Y.; Zhao, M.Y.; Du, Y.Z. Mechanisms of fungal community assembly in wild stoneflies moderated by host characteristics and local environment. NPJ Biofilms Microbiomes 2022, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.T.Y. Geographically Associated Fungus-Bacterium Interactions Contribute to the Formation of Geography-Dependent Flavor during High-Complexity Spontaneous Fermentation. Microbiol. Spectrum. 2022, 10, e0184422. [Google Scholar]
- Hu, X.; Tian, R.; Wang, K.; Cao, Z.; Yan, P.; Li, F.; Li, X.; Li, S.; He, P. The prokaryotic community, physicochemical properties and flavors dynamics and their correlations in fermented grains for Chinese strong-flavor Baijiu production. Food Res. Int. 2021, 148, 110626. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.F. Encyclopedia of Liquor Production Technology; China Light Industry Press: Beijing, China, 2007; pp. 642–653. [Google Scholar]
- NY/T 1121.7-2014; Soil Testing—Part 7: Determination of Available Phosphorus in Soil. Ministry of Agriculture: Beijing, China, 2015.
- Yuan, S.; Du, H.; Zhao, D.; Qiao, Z.; Zheng, J.; Yu, X.; Xu, Y. Stochastic Processes Drive the Assembly and Metabolite Profiles of Keystone Taxa during Chinese Strong-Flavor Baijiu Fermentation. Microbiol. Spectr. 2023, 11, e0510322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Kou, Y.; Wang, J.; Tu, B.; Li, H.; Li, X.; Wang, C.; Yao, M. Soil pH is a major driver of soil diazotrophic community assembly in Qinghai-Tibet alpine meadows. Soil Biol. Biochem. 2017, 115, 547–555. [Google Scholar] [CrossRef]
- Zhang, J. Quantitative Ecology; Science Press: Beijing, China, 2018. [Google Scholar]
- Johnson, R.; Ramond, J.B.; Gunnigle, E.; Seely, M.; Cowan, D. Deterministic Assembly of Complex Microbial Communities in Namib Desert Soils. 2016. Available online: https://www.researchgate.net/publication/309213327_Deterministic_assembly_of_complex_microbial_communities_in_Namib_Desert_soils (accessed on 3 May 2023).
- You, L.; Jian, X.; Fan, F. Ecological Monitoring of Pit Mud in Yibin Strong-fragrance Baijiuproducing Region. Biotechnol. Bull. 2023, 39, 254. [Google Scholar] [CrossRef]
- Kemmitt, S.J.; Wright, D.; Jones, D.L. Soil acidification used as a management strategy to reduce nitrate losses from agricultural land. Soil Biol. Biochem. 2005, 37, 867–875. [Google Scholar] [CrossRef]
- Kemmitt, S.; Wright, D.; Goulding, K.; Jones, D. pH regulation of carbon and nitrogen dynamics in two agricultural soils. Soil Biol. Biochem. 2006, 38, 898–911. [Google Scholar] [CrossRef]
- Shi, J.Y.; Yuan, X.F.; Lin, H.R.; Yang, Y.Q.; Li, Z.Y. Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Int. J. Mol. Sci. 2011, 12, 3770–3785. [Google Scholar] [CrossRef] [PubMed]
- Maratamutu, T. Food Safety Evaluation of Commercialize Fermented Vegetables. Agric. Food Sci. 2013. Submitted. [Google Scholar]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Baskin, C.C.; Yu, K.; Ma, Z.; Du, G. Wetland drying indirectly influences plant community and seed bank diversity through soil pH. Ecol. Indic. 2017, 80, 186–195. [Google Scholar] [CrossRef]
- Tripathi, B.M.; Kim, M.; Lai-Hoe, A.; Shukor, N.A.; Rahim, R.A.; Go, R.; Adams, J.M. pH dominates variation in tropical soil archaeal diversity and community structure. FEMS Microbiol. Ecol. 2013, 86, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Deng, J.; Xie, J. Screening of Physicochemical Quality Indexes of Pit Mud Based on Correlation with Microbial Community Structure. Food Sci. 2018, 2018, 44–50. [Google Scholar] [CrossRef]
- Mao, F.; Huang, J.; Zhou, R.; Qin, H.; Zhang, S.; Cai, X.; Qiu, C. Succession of microbial community of the pit mud under the impact of Daqu of Nongxiang Baijiu. J. Biosci. Bioeng. 2023, 136, 304–311. [Google Scholar] [CrossRef]
- Qian, W.; Lu, Z.-M.; Chai, L.J.; Zhang, X.-J.; Li, Q.; Wang, S.-T.; Shen, C.-H.; Shi, J.-S.; Xu, Z.-H. Cooperation within the microbial consortia of fermented grains and pit mud drives organic acid synthesis in strong-flavor Baijiu production. Food Res. Int. 2021, 147, 10449. [Google Scholar] [CrossRef]
- Gao, J.; Liu, G.; Li, A.; Liang, C.; Ren, C.; Xu, Y. Domination of pit mud microbes in the formation of diverse flavour compounds during Chinese strong aroma-type Baijiu fermentation. LWT 2021, 137, 10442. [Google Scholar] [CrossRef]
- Hu, X.L.; Du, H.; Xu, Y. Identification and quantification of the caproic acid-producing bacterium Clostridium kluyveri in the fermentation of pit mud used for Chinese strong-aroma type liquor production. Int. J. Food Microbiol. 2015, 214, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.J.; Xu, P.X.; Qian, W.; Zhang, X.J.; Ma, J.; Lu, Z.M.; Wang, S.T.; Shen, C.H.; Shi, J.S.; Xu, Z.H. Profiling the Clostridia with butyrate-producing potential in the mud of Chinese liquor fermentation cellar. Int. J. Food Microbiol. 2019, 297, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Dai, K.; Wang, Q.T.; Zheng, S.J.; Hong, S.D.; Jianxiong Zeng, R.; Zhang, F. Caproate production from xylose via the fatty acid biosynthesis pathway by genus Caproiciproducens dominated mixed culture fermentation. Bioresour. Technol. 2022, 351, 126978. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Feng, X.; Liang, C.; Li, J.; Jia, J.; Feng, L.; Tao, Y.; Chen, Y. Microbial Ecological Mechanism for Long-Term Production of High Concentrations of n-Caproate via Lactate-Driven Chain Elongation. Appl. Environ. Microbiol. 2021, 87, e03075-20. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shi, Z.; Luo, T.; Zhang, S.; Liu, Y.; Luo, G. Phenol promoted caproate production via two-stage batch anaerobic fermentation of organic substance with ethanol as electron donor for chain elongation. Water Res. 2021, 204, 117601. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wang, H.; Gao, J. Isolation and functional analysis of the dominant Bacteroidia species Petrimonas sulfuriphila in pit mud. Acta Microbiol. Sin. 2022, 62, 3373–3386. [Google Scholar] [CrossRef]
- Demirel, B.; Scherer, P. The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: A review. Rev. Environ. Sci. Bio/Technol. 2008, 7, 173–190. [Google Scholar] [CrossRef]
- Balch, W.E.; Fox, G.E.; Magrum, L.J.; Woese, C.R.; Wolfe, R.S. Methanogens: Reevaluation of a unique biological group. Microbiol. Rev. 1979, 43, 260–296. [Google Scholar] [CrossRef] [PubMed]
- Cintas, L.M.; Casaus, M.P.; Herranz, C.; Nes, I.F.; Hernández, P.E. Review: Bacteriocins of Lactic Acid Bacteria. Food Sci. Technol. Int. 2001, 7, 281–305. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, Y. Correlation between physicochemical factors and total bacterial count in different grades of Nongxiang flavored pit mud. Food Ferment. Ind. 2019, 45, 89–94. [Google Scholar] [CrossRef]
- Peng, Q.; Jiang, S.; Chen, J.; Ma, C.; Huo, D.; Shao, Y.; Zhang, J. Unique Microbial Diversity and Metabolic Pathway Features of Fermented Vegetables From Hainan, China. Front. Microbiol. 2018, 9, 399. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, M.; Hu, X.; Zhang, Y.; Zhang, X.; Ni, H.; Fu, D.; Chi, L. Illuminating the Characteristics and Assembly of Prokaryotic Communities across a pH Gradient in Pit Muds for the Production of Chinese Strong-Flavor Baijiu. Foods 2024, 13, 1196. https://doi.org/10.3390/foods13081196
Deng M, Hu X, Zhang Y, Zhang X, Ni H, Fu D, Chi L. Illuminating the Characteristics and Assembly of Prokaryotic Communities across a pH Gradient in Pit Muds for the Production of Chinese Strong-Flavor Baijiu. Foods. 2024; 13(8):1196. https://doi.org/10.3390/foods13081196
Chicago/Turabian StyleDeng, Mingdong, Xiaolong Hu, Yong Zhang, Xinyu Zhang, Haifeng Ni, Danyang Fu, and Lei Chi. 2024. "Illuminating the Characteristics and Assembly of Prokaryotic Communities across a pH Gradient in Pit Muds for the Production of Chinese Strong-Flavor Baijiu" Foods 13, no. 8: 1196. https://doi.org/10.3390/foods13081196