Changes in Protein Non-Covalent Bonds and Aggregate Size during Dough Formation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Wheat Flour
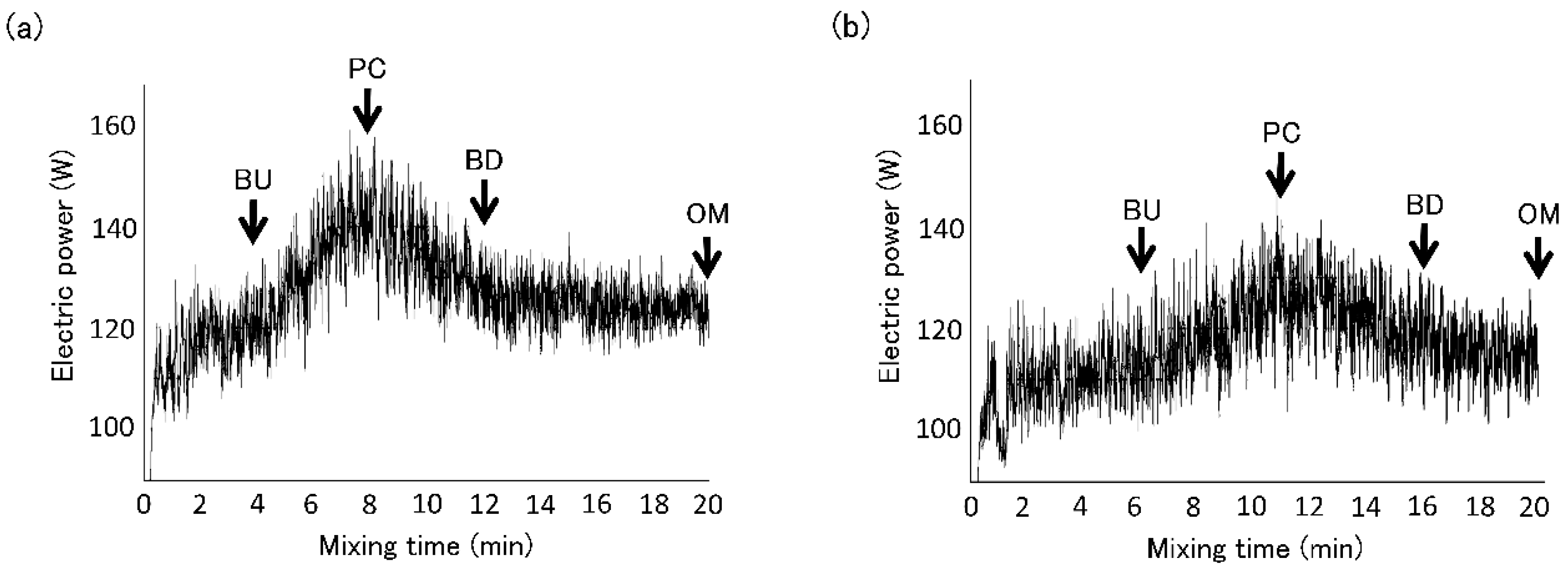
2.2. Dough Sampling during Mixing
2.3. Protein Extraction
2.4. Analysis of Protein Size Distribution by SE-HPLC
2.5. Analysis of Protein Size Distribution by FFF
2.6. Identification of Protein by 2Dimensional Fluorescence Difference Gel Electrophoresis (2D-DIGE)
2.7. Statistics
3. Results
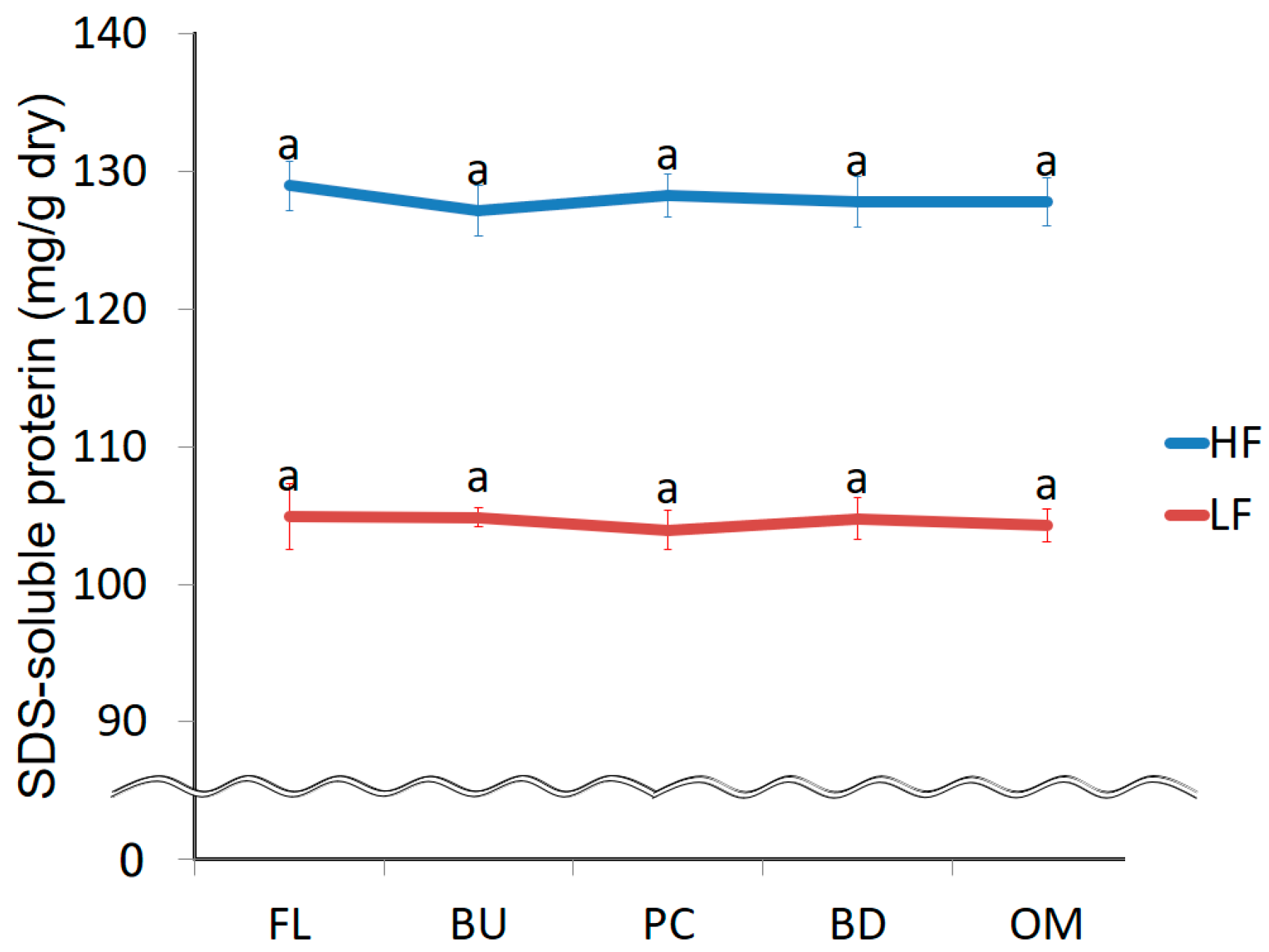
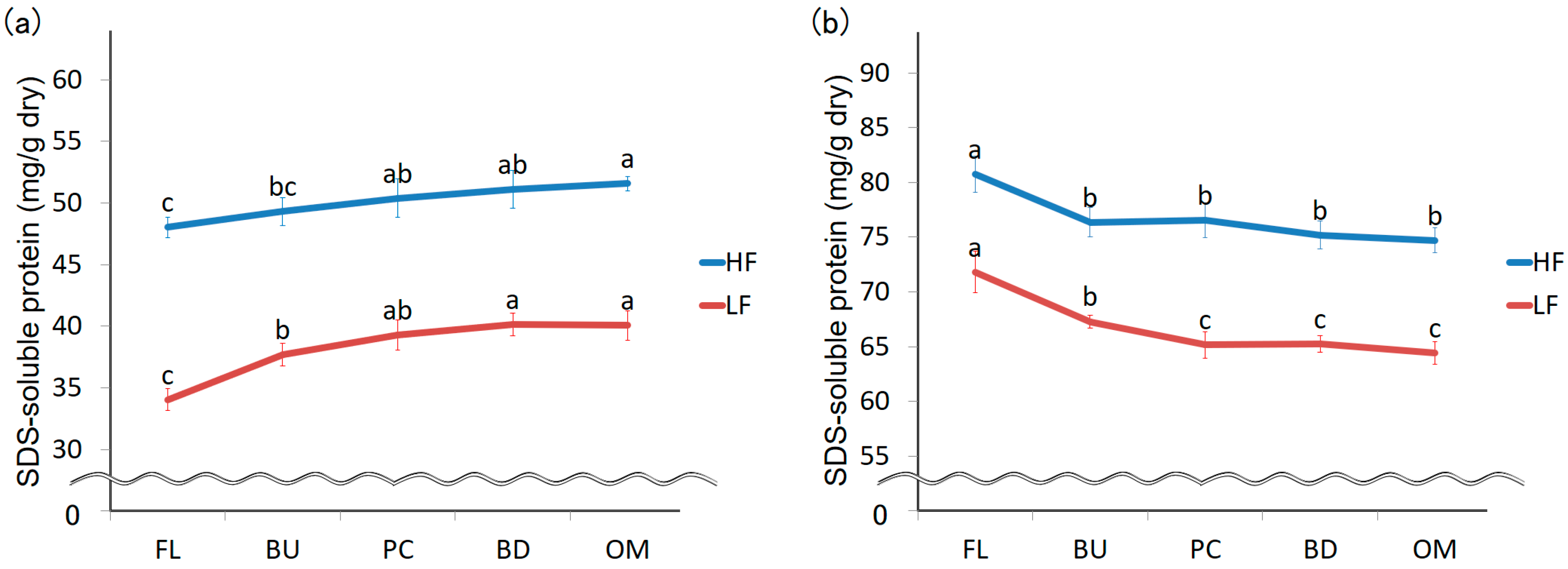
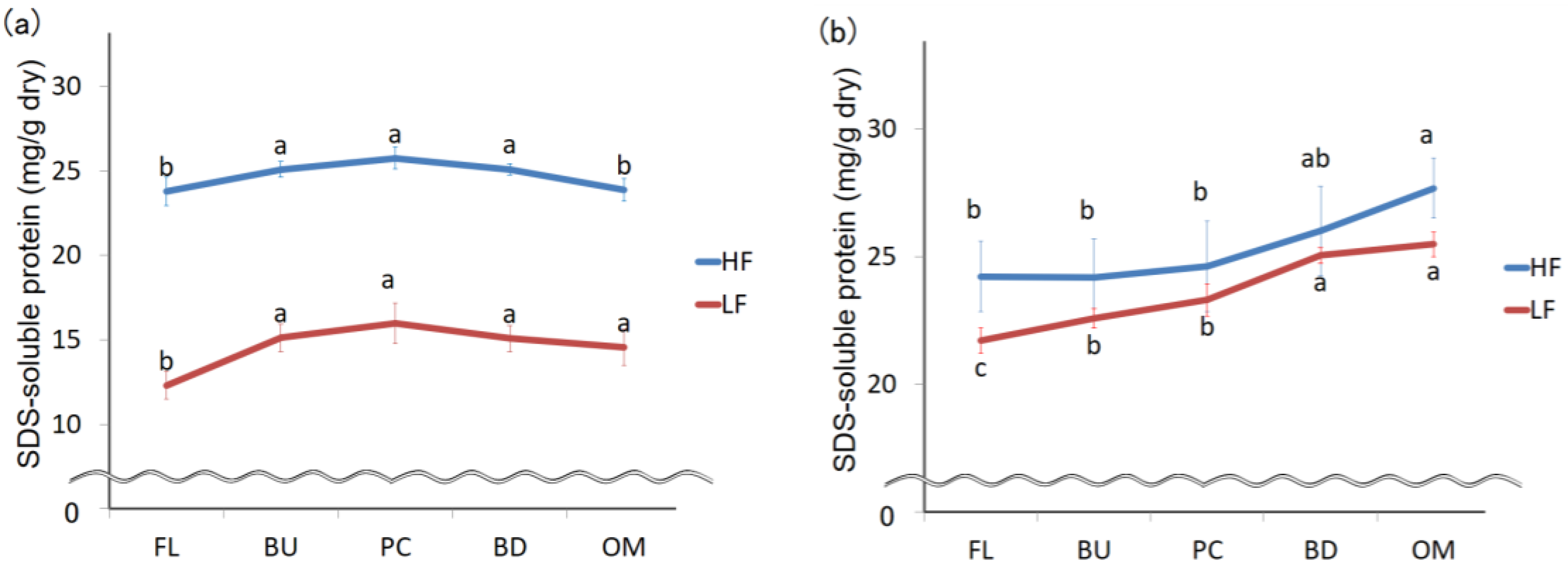
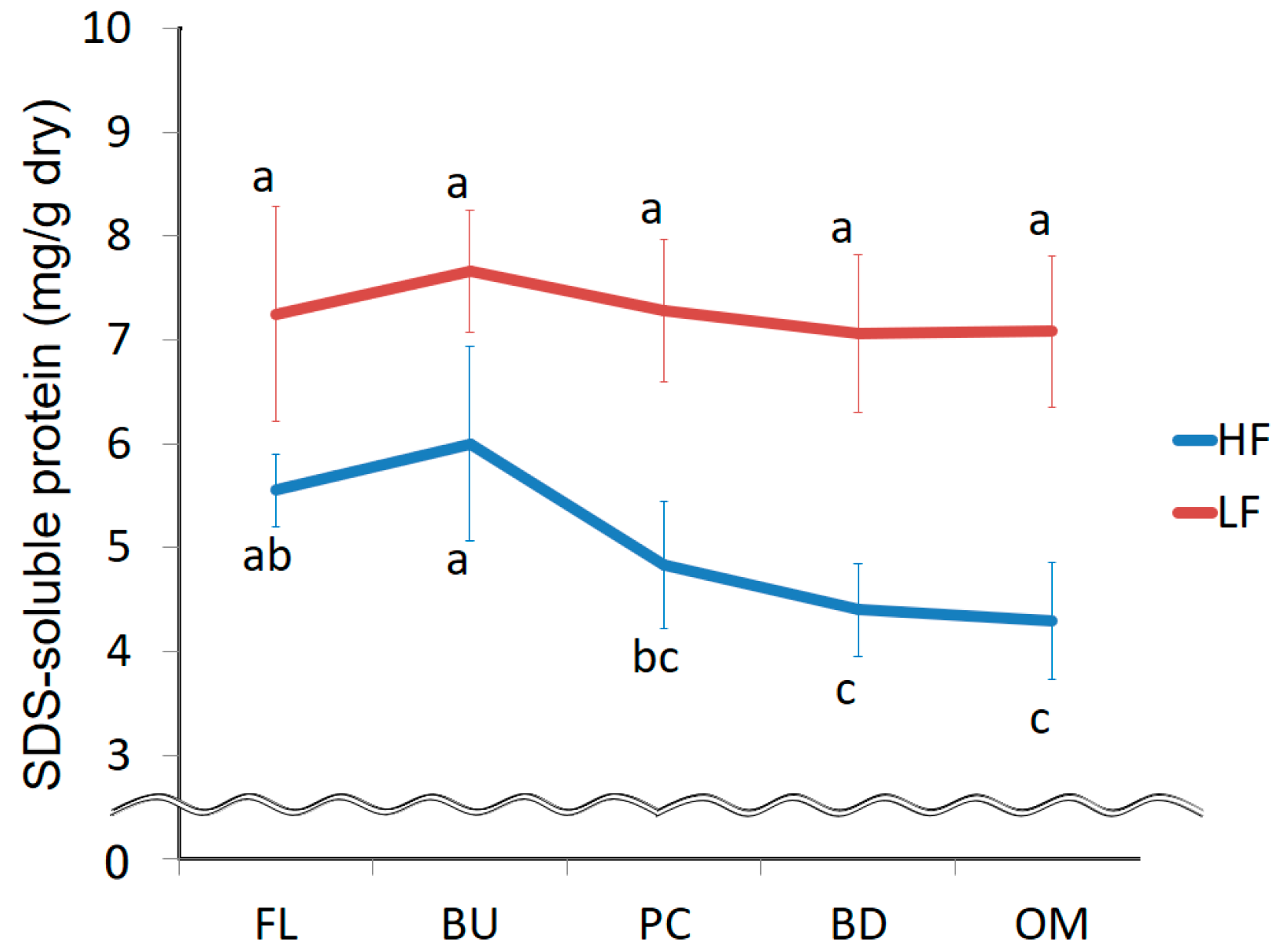
3.1. Changes in the Amount of Extracted Protein during Mixing
3.2. Change in Protein Size Distribution during Mixing
3.3. Change in the Largest Protein Size Fraction during Mixing
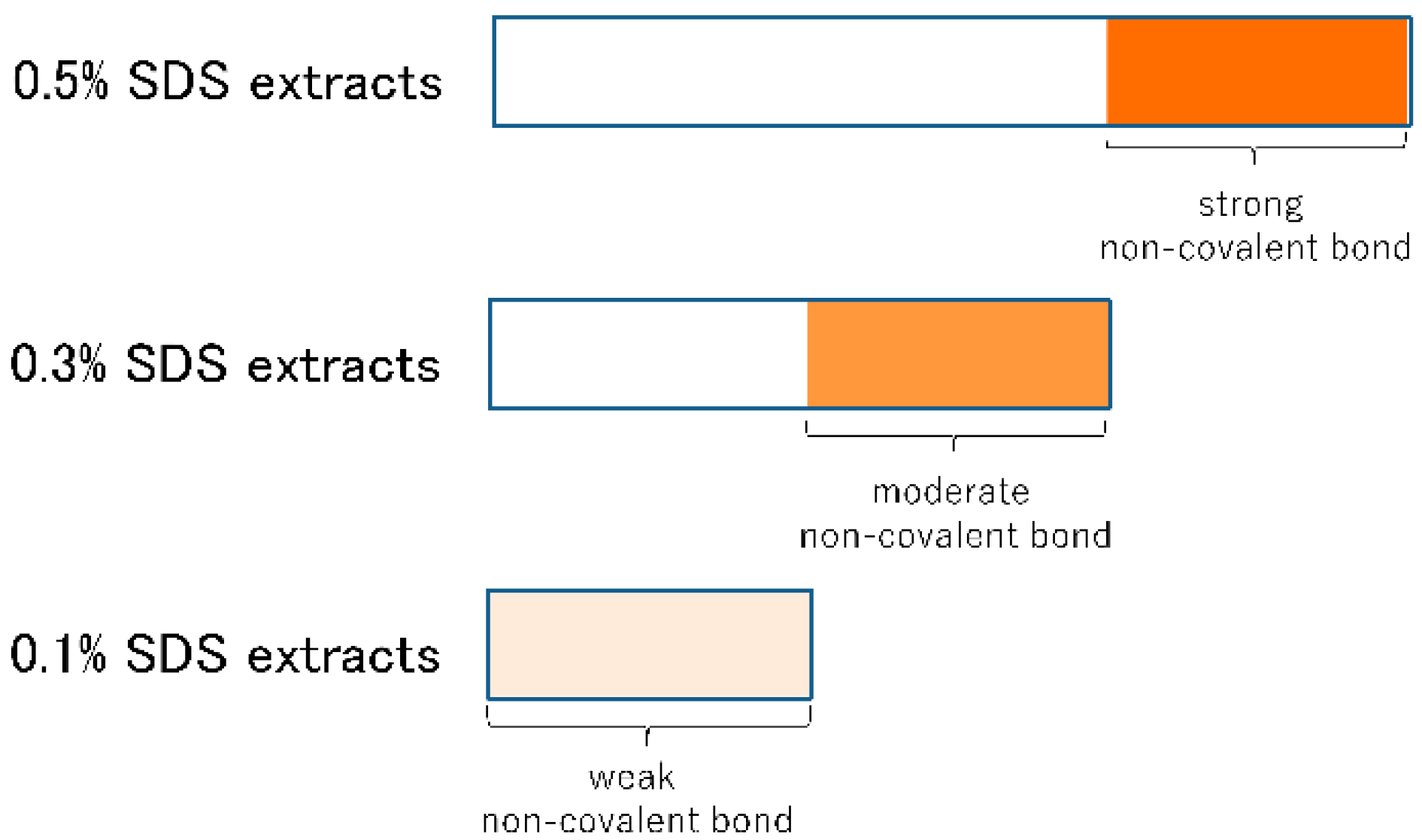
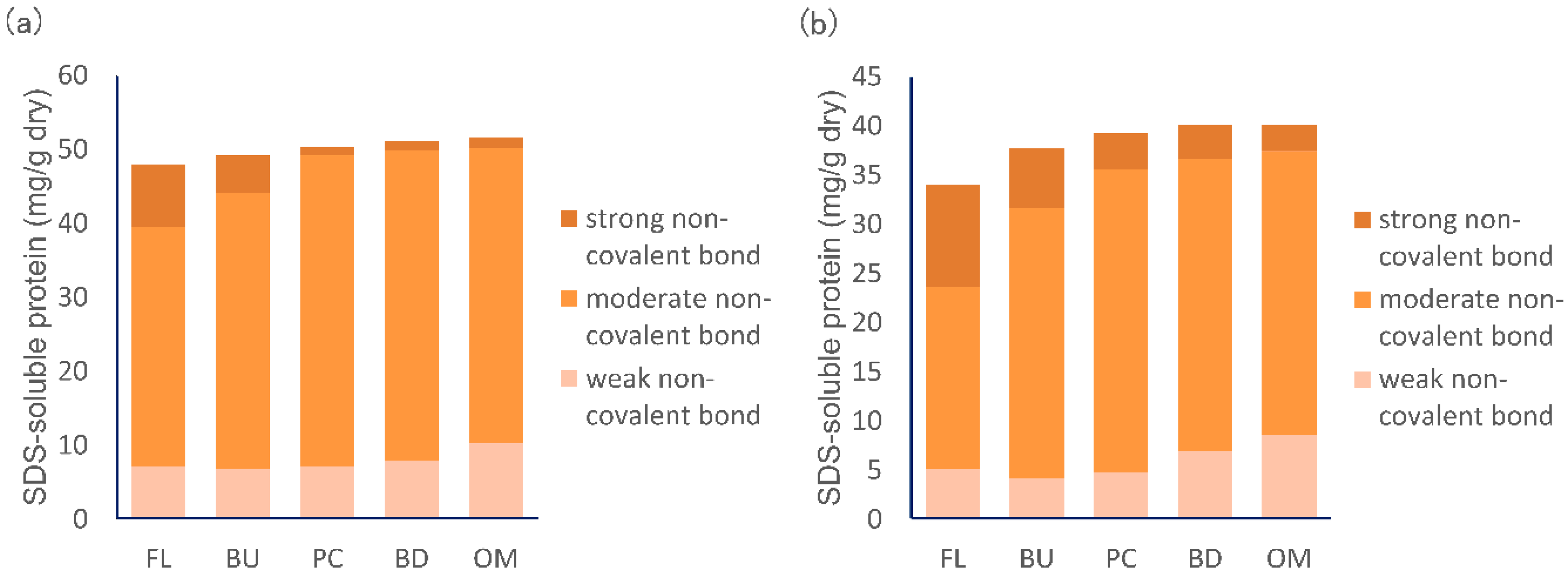
3.4. Change in the Strength of Non-Covalent Bonds during Mixing
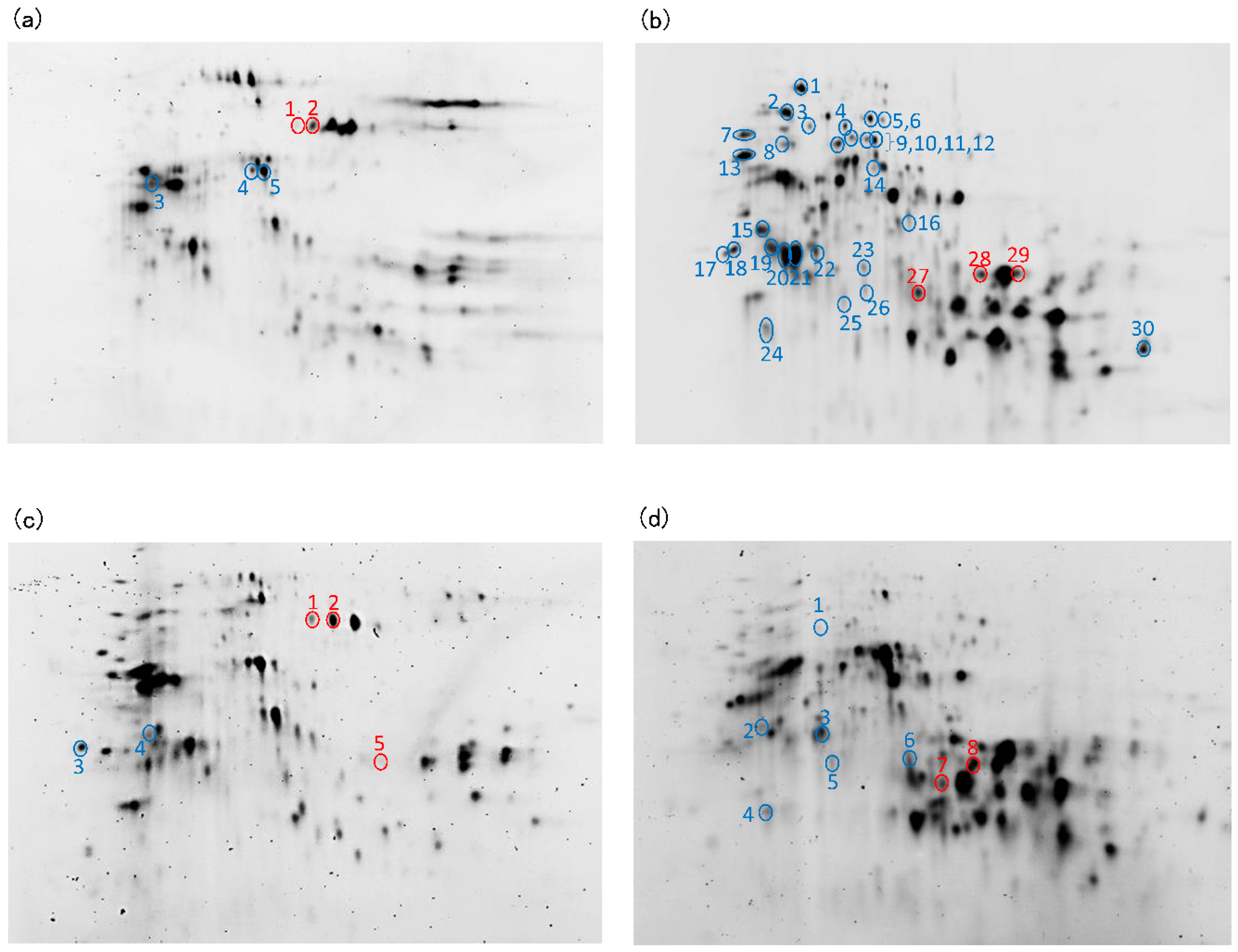
3.5. Identification of Monomeric Protein Incorporated in Protein Aggregates during Mixing
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Kasarda, D.D. Glutenin structure in relation to wheat quality. In Wheat Is Unique; Pomeranz, Y., Ed.; AACC International Press: St. Paul, MN, USA, 1989; pp. 277–302. [Google Scholar]
- Wrigley, C.W. Giant proteins with flour power. Nature 1996, 381, 738–739. [Google Scholar] [CrossRef]
- Wieser, H.; Bushuk, W.; MacRitchie, F. The polymeric glutenins. In Gliadin and Glutenin: The Unique Balance of Wheat Quality; Wrigley, C., Bekes, F., Bushuk, W., Eds.; AACC International Press: St. Paul, MN, USA, 2006; pp. 213–240. [Google Scholar]
- Tuhumury, H.C.D.; Small, D.M.; Day, L. The effect of sodium chloride on gluten network formation and rheology. J. Cereal Sci. 2014, 60, 229–237. [Google Scholar] [CrossRef]
- Weegels, P.L.; Marseille, J.P.; Jager, A.M.; Hamer, R.J. Structure-function relationships of gluten proteins. In Gluten Proteins; Bushuk, W., Tkachuk, R., Eds.; AACC International Press: St. Paul, MN, USA, 1990; pp. 98–111. [Google Scholar]
- Gao, L.; Ng, P.K.W.; Bushuk, W. Structure of glutenin based on farinograph and electrophoretic results. Cereal Chem. 1992, 69, 452–455. [Google Scholar]
- MacRitchie, F. Letters to the editor. J. Cereal Sci. 2007, 60, 96–97. [Google Scholar] [CrossRef]
- MacRitchie, F. Theories of glutenin/dough systems. J. Cereal Sci. 2014, 60, 4–6. [Google Scholar] [CrossRef]
- Belton, P. Letters to the editor. J. Cereal Sci. 2007, 60, 97–98. [Google Scholar] [CrossRef]
- van Vliet, T.; Hamer, R.J. Letters to the editor. J. Cereal Sci. 2007, 60, 98–99. [Google Scholar] [CrossRef]
- Delcour, J.A.; Joye, I.J.; Pareyt, B.; Wilderjans, E.; Brijs, K.; Lagrain, K. Wheat gluten functionality as a quality determinant in cereal-based food products. Annu. Rev. Food Sci. Technol. 2012, 3, 469–492. [Google Scholar] [CrossRef]
- Shewry, P.R.; D’Ovid, R.; Lafiandra, R.D.D.; Jenkins, J.A.; Mills, E.N.C.; Bekes, F. Wheat Grain Proteins. In Wheat: Chemistry and Technology, 4th ed.; Khan, K., Shewry, P.R., Eds.; AACC International Press: St. Paul, MN, USA, 2009; pp. 223–298. [Google Scholar]
- Kinsella, J.E.; Hale, M.L. Hydrophobic associations and gluten consistency: Effects of specific anions. J. Agric. Food Chem. 1984, 32, 1054–1056. [Google Scholar] [CrossRef]
- Miller, R.A.; Hoseney, R.C. Role of salt in baking. Cereal Foods World 2008, 53, 4–6. [Google Scholar] [CrossRef]
- Belton, P.S. On the elasticity of wheat gluten. J. Cereal Sci. 1999, 29, 103–107. [Google Scholar] [CrossRef]
- van Velzen, E.J.J.; van Duynhoven, J.P.M.; Pudney, P.; Weegels, P.L.; van der Maas, J.H. Factors associated with dough stickiness as sensed by attenuated total reflectance infrared spectroscopy. Cereal Chem. J. 2003, 80, 378–382. [Google Scholar] [CrossRef]
- Bock, J.E.; West, R.; Iametti, S.; Bonomi, F.; Marengo, M.; Seetharaman, K. Gluten structural evolution during pasta processing of refined and whole wheat pasta from hard white winter wheat: The influence of mixing, drying, and cooking. Cereal Chem. J. 2015, 92, 460–465. [Google Scholar] [CrossRef]
- Jazaeri, S.; Bock, J.E.; Bagagli, M.P.; Iametti, S.; Bonomi, F.; Seetharaman, K. Structural modifications of gluten proteins in strong and weak wheat dough during mixing. Cereal Chem. J. 2015, 92, 105–113. [Google Scholar] [CrossRef]
- Bonomi, F.; Mora, G.; Pagani, M.A.; Iametti, S. Probing structural features of water-insoluble proteins by front-face fluorescence. Anal. Biochem. 2004, 329, 104–111. [Google Scholar] [CrossRef]
- Kuktaite, R.; Larsson, H.; Johansson, E. Variation in protein composition of wheat flour and its relationship to dough mixing behavior. J. Cereal Sci. 2004, 40, 31–39. [Google Scholar] [CrossRef]
- Singh, N.K.; Donovan, G.R.; Batey, I.L.; MacRitchie, F. Use of sonication and size-exclusion high-performance liquid chromatography in the study of wheat flour proteins. I. Dissolution of total proteins in the absence of reducing agents. Cereal Chem. 1990, 67, 150–161. [Google Scholar]
- Hamer, R.J.; MacRitchie, F.; Weegels, P.L. Structure and Functional Properties of Gluten. In Wheat: Chemistry and Technology, 4th ed.; Khan, K., Shewry, P.R., Eds.; AACC International Press: St. Paul, MN, USA, 2009; pp. 153–178. [Google Scholar]
- Tanaka, K.; Bushuk, W. Changes in flour proteins during dough-mixing. I. Solubility results. Cereal Chem. 1973, 50, 590–596. [Google Scholar]
- MacRitchie, F. Mechanical degradation of gluten proteins during high-speed mixing of doughs. J. Polym. Sci. Symp. 1975, 49, 85–90. [Google Scholar] [CrossRef]
- Danno, G.; Hoseney, R.C. Changes in flour proteins during dough mixing. Cereal Chem. 1982, 59, 249–253. [Google Scholar]
- Dupont, F.M.; Vensel, W.H.; Tanaka, C.K.; Hurkman, W.J.; Altenbach, S.B. Deciphering the complexities of the wheat flour proteome using quantitative two-dimensional electrophoresis, three proteases and tandem mass spectropetry. Proteome Sci. 2011, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecham, D.K.; Cole, E.G.; Pence, J.W. Dough-mixing properties of crude and purified glutens. Cereal Chem. 1965, 42, 409–420. [Google Scholar]
- Tsen, C.C. Changes in flour proteins during dough mixing. Cereal Chem. 1967, 44, 308–316. [Google Scholar]
- Eckert, B.; Amend, T.; Belitz, H.D. The course of the SDS and Zeleny sedimentation tests for gluten quality and related phenomena studied using light-microscope. Z. Lebensm. Unters. Forsch. 1993, 196, 122–125. [Google Scholar] [CrossRef]
- Belton, P.S. New approaches to study the molecular basis of the mechanical properties of gluten. J. Cereal Sci. 2005, 41, 203–211. [Google Scholar] [CrossRef]
- Bueche, F. Molecular basis for mullins effect. J. Appl. Polym. Sci. 1960, 4, 107–114. [Google Scholar] [CrossRef]
- Aussenac, T.; Carceller, J.L.; Kleiber, D. Changes in SDS solubility of gluten polymers during dough mixing and resting. Cereal Chem. 2001, 78, 39–45. [Google Scholar] [CrossRef]
- Sievert, D.; Sapirstein, H.D.; Bushuk, W. Changes in electrophoretic patterns of acetic acid-insoluble wheat flour proteins during dough mixing. J. Cereal Sci. 1991, 14, 243–256. [Google Scholar] [CrossRef]
- Skerritt, J.H.; Hac, L.; Bekes, F. Depolymerization of the glutenin macropolymer during dough mixing: I. Changes in levels, molecular weight distribution, and overall composition. Cereal Chem. 1999, 76, 395–401. [Google Scholar] [CrossRef]
- Skerritt, J.H.; Hac, L.; Lindsay, M.P.; Bekes, F. Depolymerization of the glutenin macropolymer during dough mixing: II. Differences in retention of specific glutenin subunits. Cereal Chem. 1999, 76, 402–409. [Google Scholar] [CrossRef]
- La Gatta, B.; Rutigliano, M.; Rusco, G.; Petrella, G.; Di Luccia, A. Evidence for different supramolecular arrangements in pasta from durum wheat (Triticum durum) and einkorn (Triticum monococcum) flours. J. Cereal Sci. 2017, 73, 76–83. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot No. b | Identification c | Spot Ratio (%) d | Increase (↑) or Decrease (↓) | ||||
|---|---|---|---|---|---|---|---|
| Flour | Build up | Peak Consistency | Break Down | Overmixing | |||
| 1 | HMW-glutenin | 0.52 ± 0.25 | 0.64 ± 0.07 | 1.04 ± 0.09 | 1.12 ± 0.27 | 1.19 ± 0.17 | ↑ |
| 2 | HMW-glutenin | 0.59 ± 0.15 | 0.75 ± 0.05 | 1.17 ± 0.22 | 1.32 ± 0.27 | 1.36 ± 0.13 | ↑ |
| 3 | ω-gliadin | 1.12 ± 0.55 | 1.72 ± 0.54 | 0.74 ± 0.1 | 0.72 ± 0.24 | 0.85 ± 0.25 | ↓ |
| 4 | ω-gliadin | 1.66 ± 0.19 | 1.24 ± 0.18 | 0.73 ± 0.62 | 0.66 ± 0.38 | 0.56 ± 0.14 | ↓ |
| 5 | ω-gliadin | 2.08 ± 1.13 | 1.14 ± 0.34 | 0.69 ± 0.15 | 0.73 ± 0.13 | 0.62 ± 0.16 | ↓ |
| Spot No. b | Identification c | Spot Ratio (%) d | Increase (↑) or Decrease (↓) | ||||
|---|---|---|---|---|---|---|---|
| Flour | Build up | Peak Consistency | Break Down | Overmixing | |||
| 1 | Enzymes | 1.82 ± 0.39 | 0.51 ± 0.19 | 0.56 ± 0.15 | 0.46 ± 0.45 | 0.95 ± 0.91 | ↓ |
| 2 | Enzymes | 1.88 ± 0.35 | 0.54 ± 0.22 | 0.65 ± 0.21 | 0.42 ± 0.42 | 0.8 ± 0.38 | ↓ |
| 3 | Enzymes | 1.54 ± 0.87 | 0.44 ± 0.1 | 0.56 ± 0.11 | 0.46 ± 0.3 | 0.63 ± 0.27 | ↓ |
| 4 | Enzymes | 2 ± 1.22 | 0.57 ± 0.52 | 0.71 ± 0.38 | 0.45 ± 0.11 | 0.55 ± 0.13 | ↓ |
| 5 | Enzymes | 1.83 ± 0.88 | 0.39 ± 0.16 | 0.59 ± 0.16 | 0.4 ± 0.28 | 0.58 ± 0.53 | ↓ |
| 6 | Enzymes | 1.76 ± 0.83 | 0.23 ± 0.22 | 0.54 ± 0.26 | 0.41 ± 0.12 | 0.5 ± 0.29 | ↓ |
| 7 | Enzymes | 2.04 ± 0.79 | 0.56 ± 0.56 | 0.64 ± 0.29 | 0.35 ± 0.2 | 0.42 ± 0.34 | ↓ |
| 8 | Enzymes | 1.66 ± 0.42 | 0.74 ± 0.36 | 0.74 ± 0.19 | 0.46 ± 0.12 | 0.66 ± 0.36 | ↓ |
| 9 | Enzymes | 2.05 ± 0.98 | 0.53 ± 0.21 | 0.61 ± 0.21 | 0.52 ± 0.3 | 0.61 ± 0.3 | ↓ |
| 10 | Enzymes | 1.95 ± 1.19 | 0.67 ± 0.38 | 0.8 ± 0.39 | 0.54 ± 0.07 | 0.65 ± 0.23 | ↓ |
| 11 | Enzymes | 1.89 ± 1.1 | 0.48 ± 0.04 | 0.48 ± 0.18 | 0.33 ± 0.22 | 0.41 ± 0.37 | ↓ |
| 12 | Enzymes | 1.87 ± 0.7 | 0.5 ± 0.17 | 0.45 ± 0.25 | 0.37 ± 0.25 | 0.58 ± 0.77 | ↓ |
| 13 | Enzymes | 2.53 ± 1.07 | 0.64 ± 0.35 | 0.54 ± 0.27 | 0.35 ± 0.21 | 0.45 ± 0.45 | ↓ |
| 14 | Enzymes | 1.21 ± 0.32 | 0.76 ± 0.21 | 1.01 ± 0.34 | 0.49 ± 0.25 | 0.38 ± 0.18 | ↓ |
| 15 | ω-gliadin | 1.77 ± 0.73 | 0.93 ± 1.07 | 0.71 ± 0.16 | 0.41 ± 0.12 | 0.6 ± 0.28 | ↓ |
| 16 | ω-gliadin | 1.56 ± 0.33 | 0.82 ± 0.25 | 0.86 ± 0.04 | 0.71 ± 0.24 | 0.73 ± 0.3 | ↓ |
| 17 | ω-gliadin | 1.76 ± 0.34 | 0.65 ± 0.07 | 0.73 ± 0.11 | 0.62 ± 0.16 | 0.76 ± 0.26 | ↓ |
| 18 | ω-gliadin | 1.9 ± 0.37 | 0.78 ± 0.17 | 0.86 ± 0.13 | 0.62 ± 0.11 | 0.9 ± 0.33 | ↓ |
| 19 | ω-gliadin | 1.56 ± 0.47 | 0.44 ± 0.17 | 0.76 ± 0.23 | 0.52 ± 0.22 | 0.53 ± 0.29 | ↓ |
| 20 | ω-gliadin | 2.39 ± 1.15 | 0.7 ± 0.26 | 0.78 ± 0.17 | 0.51 ± 0.2 | 0.91 ± 0.16 | ↓ |
| 21 | ω-gliadin | 2.85 ± 0.99 | 0.63 ± 0.64 | 0.6 ± 0.2 | 0.38 ± 0.25 | 0.88 ± 0.76 | ↓ |
| 22 | ω-gliadin | 1.64 ± 0.35 | 0.6 ± 0.15 | 0.64 ± 0.17 | 0.75 ± 0.22 | 0.75 ± 0.34 | ↓ |
| 23 | ω-gliadin | 1.72 ± 1.06 | 0.47 ± 0.08 | 0.68 ± 0.28 | 0.51 ± 0.1 | 0.64 ± 0.41 | ↓ |
| 24 | Serpins | 2.28 ± 0.68 | 0.59 ± 0.15 | 0.8 ± 0.3 | 0.55 ± 0.17 | 0.53 ± 0.41 | ↓ |
| 25 | Serpins | 1.64 ± 0.59 | 0.74 ± 0.15 | 0.88 ± 0.15 | 0.68 ± 0.08 | 0.82 ± 0.24 | ↓ |
| 26 | Serpins | 1.7 ± 0.81 | 0.51 ± 0.09 | 0.79 ± 0.39 | 0.67 ± 0.12 | 0.71 ± 0.14 | ↓ |
| 27 | α-gliadin | 0.68 ± 0.19 | 1.47 ± 0.21 | 0.97 ± 0.16 | 1.18 ± 0.19 | 1.42 ± 0.65 | ↑ |
| 28 | LMW-glutenin | 0.78 ± 0.12 | 1.16 ± 0.09 | 1.16 ± 0.26 | 1.57 ± 0.52 | 1.41 ± 0.4 | ↑ |
| 29 | LMW-glutenin | 0.8 ± 0.11 | 1.21 ± 0.24 | 1.17 ± 0.1 | 1.63 ± 0.81 | 1.49 ± 0.3 | ↑ |
| 30 | Inhibitors | 1.32 ± 0.27 | 0.56 ± 0.25 | 0.54 ± 0.25 | 0.6 ± 0.16 | 0.51 ± 0.02 | ↓ |
| Spot No. b | Identification c | Spot Ratio (%) d | Increase (↑) or Decrease (↓) | ||||
|---|---|---|---|---|---|---|---|
| Flour | Build up | Peak Consistency | Break Down | Overmixing | |||
| 1 | HMW-glutenin | 0.7 ± 0.15 | 0.89 ± 0.1 | 1.51 ± 0.38 | 1.64 ± 0.17 | 1.8 ± 0.74 | ↑ |
| 2 | HMW-glutenin | 0.76 ± 0.19 | 1.09 ± 0.21 | 1.57 ± 0.45 | 1.47 ± 0.11 | 1.48 ± 0.64 | ↑ |
| 3 | ω-gliadin | 0.7 ± 0.16 | 0.81 ± 0.23 | 1 ± 0.37 | 1.94 ± 0.71 | 2.2 ± 1.2 | ↓ |
| 4 | ω-gliadin | 1.6 ± 0.37 | 1.35 ± 0.27 | 0.73 ± 0.07 | 1.11 ± 0.37 | 1.1 ± 0.2 | ↓ |
| 5 | LMW-glutenin | 0.7 ± 0.21 | 1.13 ± 0.21 | 1.6 ± 0.64 | 1.33 ± 0.21 | 1.3 ± 0.52 | ↑ |
| Spot No. b | Identificationc | Spot Ratio (%) d | Increase (↑) or Decrease (↓) | ||||
|---|---|---|---|---|---|---|---|
| Flour | Build up | Peak Consistency | Break Down | Overmixing | |||
| 1 | Enzymes | 1.52 ± 0.58 | 0.48 ± 0.08 | 0.47 ± 0.2 | 0.35 ± 0.24 | 0.34 ± 0.27 | ↓ |
| 2 | ω-gliadin | 2.16 ± 0.8 | 0.88 ± 0.55 | 0.67 ± 0.1 | 0.55 ± 0.19 | 0.45 ± 0.24 | ↓ |
| 3 | ω-gliadin | 1.33 ± 0.16 | 0.87 ± 0.16 | 0.85 ± 0.07 | 0.88 ± 0.13 | 0.91 ± 0.1 | ↓ |
| 4 | Serpins | 2.48 ± 0.52 | 0.6 ± 0.12 | 0.48 ± 0.09 | 0.43 ± 0.22 | 0.48 ± 0.21 | ↓ |
| 5 | Serpins | 1.6 ± 0.22 | 0.74 ± 0.13 | 0.82 ± 0.1 | 0.83 ± 0.18 | 0.88 ± 0.33 | ↓ |
| 6 | Enzymes | 2.07 ± 0.42 | 1.21 ± 0.15 | 1.09 ± 0.42 | 0.88 ± 0.22 | 0.99 ± 0.25 | ↓ |
| 7 | α-gliadin | 0.63 ± 0.14 | 0.88 ± 0.24 | 1.21 ± 0.29 | 1.34 ± 0.16 | 1.53 ± 0.22 | ↑ |
| 8 | γ-gliadin | 0.84 ± 0.16 | 1.28 ± 0.23 | 1.52 ± 0.28 | 1.41 ± 0.07 | 1.56 ± 0.41 | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwaki, S.; Aono, S.; Hayakawa, K.; Fu, B.X.; Otobe, C. Changes in Protein Non-Covalent Bonds and Aggregate Size during Dough Formation. Foods 2020, 9, 1643. https://doi.org/10.3390/foods9111643
Iwaki S, Aono S, Hayakawa K, Fu BX, Otobe C. Changes in Protein Non-Covalent Bonds and Aggregate Size during Dough Formation. Foods. 2020; 9(11):1643. https://doi.org/10.3390/foods9111643
Chicago/Turabian StyleIwaki, Sonoo, Shiro Aono, Katsuyuki Hayakawa, Bin Xiao Fu, and Chikako Otobe. 2020. "Changes in Protein Non-Covalent Bonds and Aggregate Size during Dough Formation" Foods 9, no. 11: 1643. https://doi.org/10.3390/foods9111643
APA StyleIwaki, S., Aono, S., Hayakawa, K., Fu, B. X., & Otobe, C. (2020). Changes in Protein Non-Covalent Bonds and Aggregate Size during Dough Formation. Foods, 9(11), 1643. https://doi.org/10.3390/foods9111643