Quantitative Determination of Acrolein in Cider by 1H NMR Spectrometry


Abstract
:
1. Introduction
2. Materials and Methods
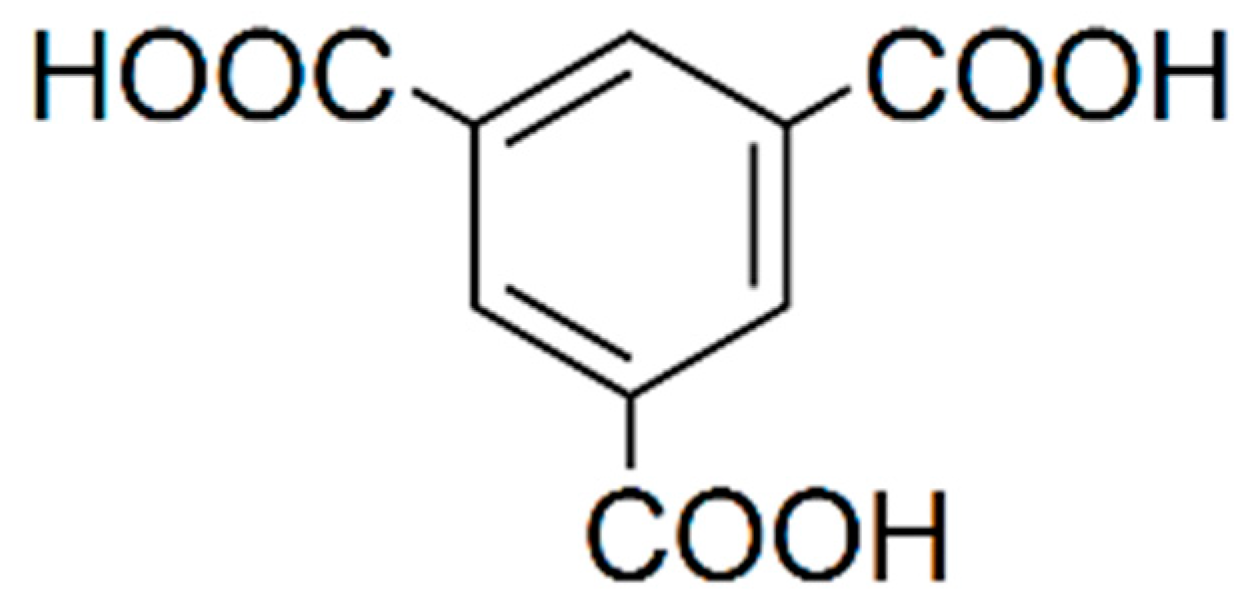
2.1. Chemicals and Reagents
2.2. Preparing the TSP-BTC-D2O Solution
2.3. Recording of 1H NMR Spectra: General Procedure
2.4. Preparation of Stock Solution of Acrolein
2.5. Calibration Graph
2.6. Determination of the Longitudinal Relaxation Time, T1
2.7. Preparing the Cider Samples
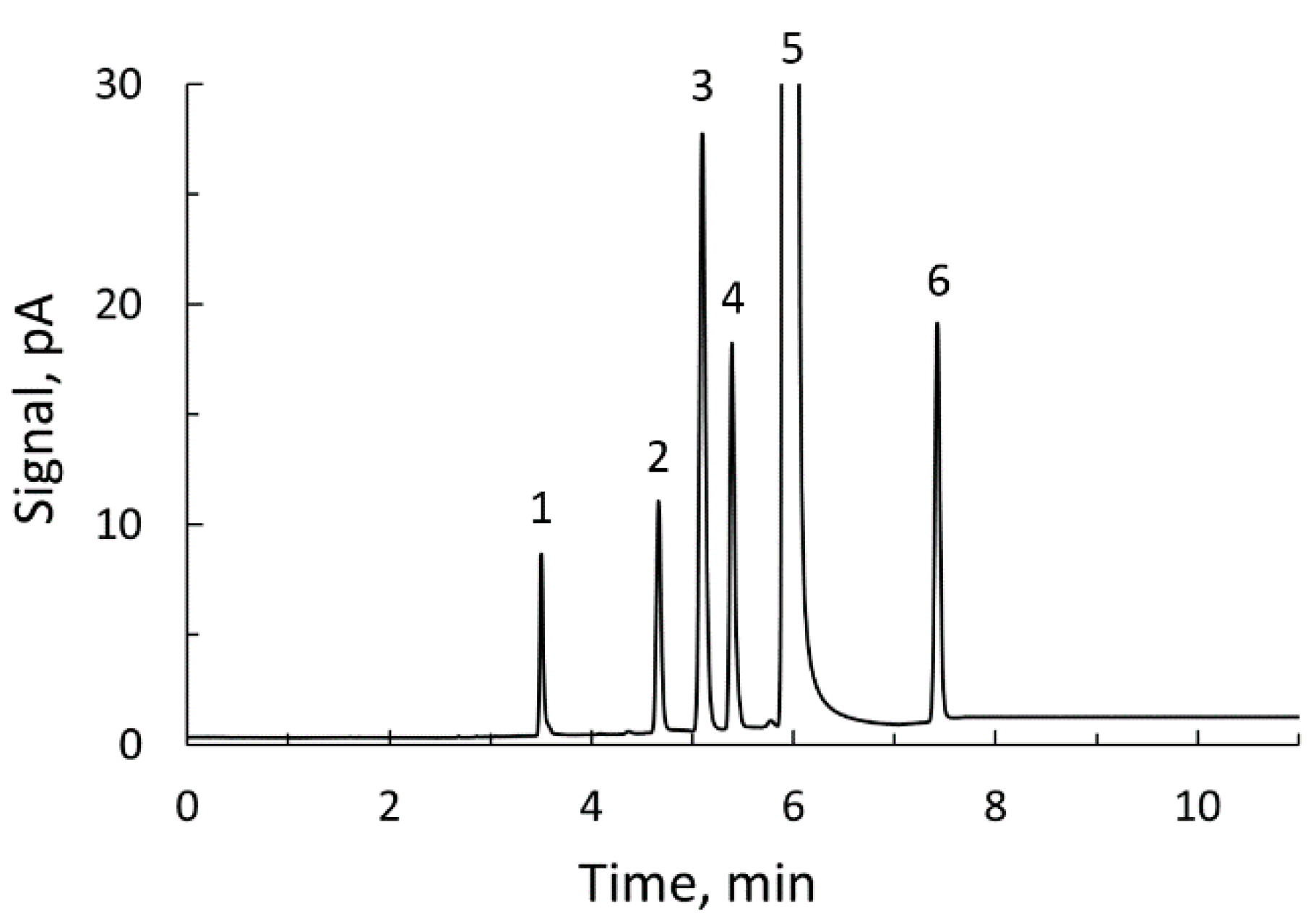
2.8. Analysis of Samples by Gas Chromatography
3. Results
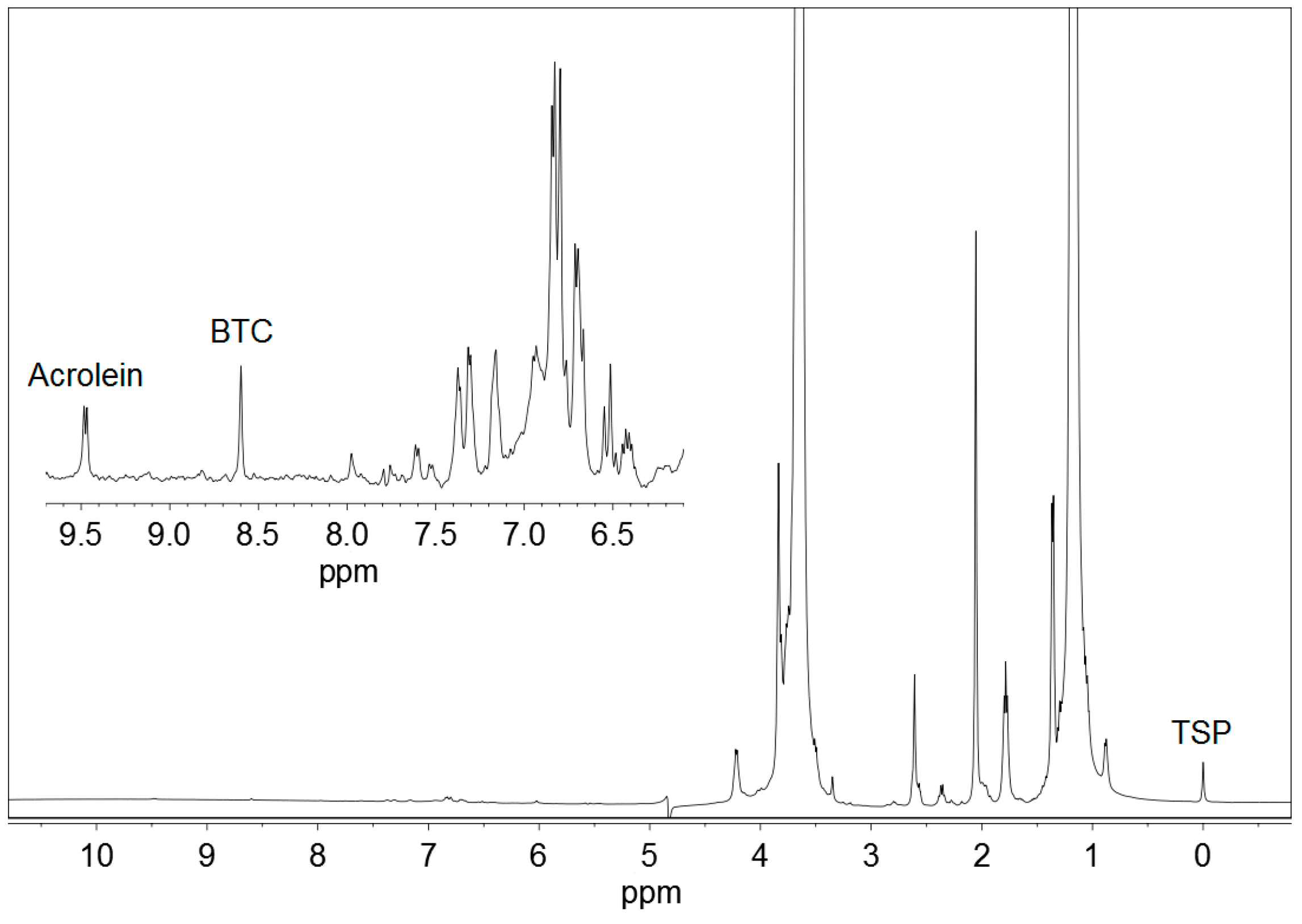
3.1. H NMR Spectra of Ciders
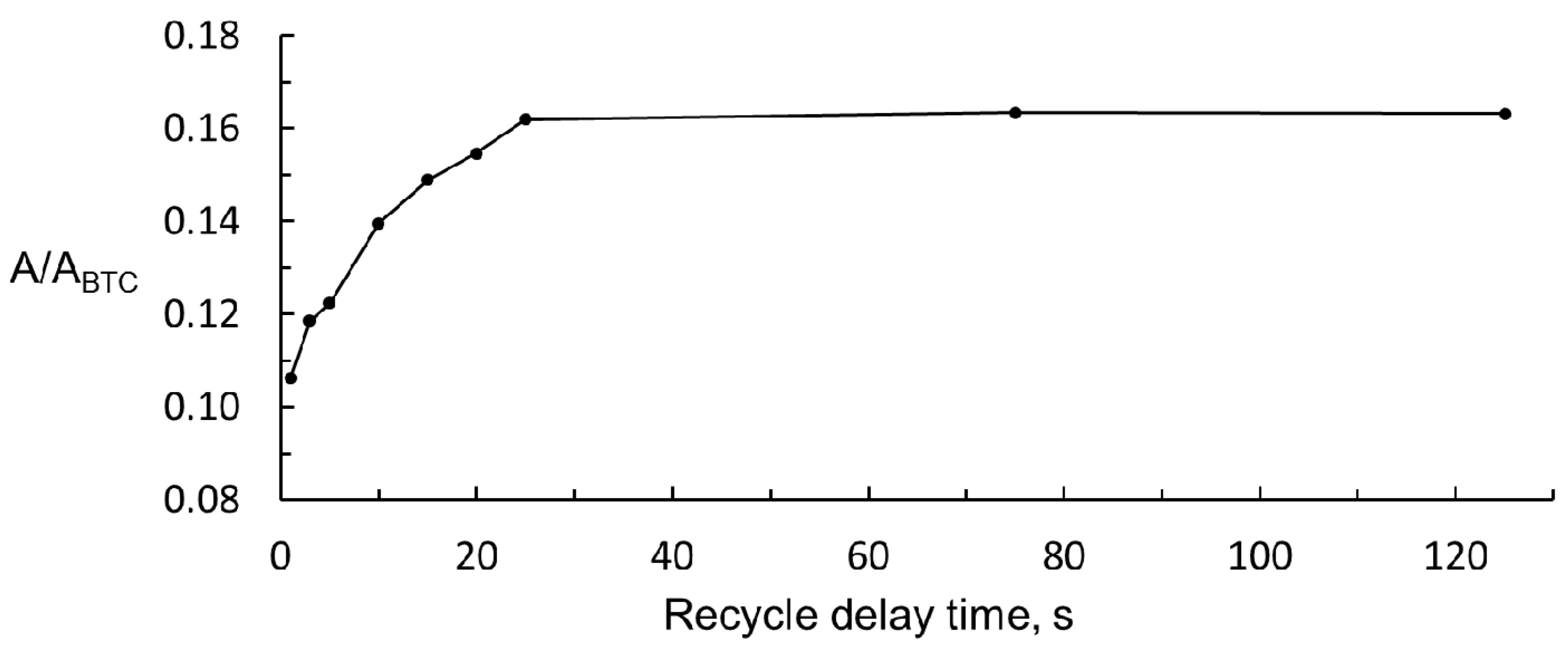
3.2. Optimisation of 1H NMR Acquisition Conditions
3.3. Calibration Equation and Limit of Detection
3.4. Precision
3.5. Application to Commercial Ciders
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gomes, R.; Meek, M.E.; Eggleton, M. Acrolein. Concise International Chemical Assessment Document 43; World Health Organization: Geneva, Switzerland, 2002. [Google Scholar]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malondialdehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Vollenweider, S.; Lacroix, C. 3-Hydroxypropionaldehyde: Applications and perspectives of biotechnological production. Appl. Microbiol. Biotechnol. 2004, 64, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Garai-Ibabe, G.; Ibarburu, I.; Berregi, I.; Claisse, O.; Lonvaud-Funel, A.; Irastorza, A.; Dueñas, M.T. Glycerol metabolism and bitterness producing lactic acid bacteria in cidermaking. Int. J. Food Microbiol. 2008, 121, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Warcollier, G.; Le Moal, A. Accidental presence of acrolein in cider brandy. Ann. Falsif. Fraud. 1932, 25, 271–273. [Google Scholar]
- Claisse, O.; Lonvaud-Funel, A. Assimilation of glycerol by a strain of Lactobacillus collinoides isolated from cider. Food Microbiol. 2000, 17, 513–519. [Google Scholar] [CrossRef]
- Sauvageot, N.; Gouffi, K.; Laplace, J.M.; Auffray, Y. Glycerol metabolism in Lactobacillus collinoides: Production of 3-hydroxypropionaldehyde, a precursor of acrolein. Int. J. Food Microbiol. 2000, 55, 167–170. [Google Scholar] [CrossRef]
- Oliveira Lago, L.; Primieri Nicolli, K.; Biasoto Marques, A.; Alcaraz Zini, C.; Elisa Welke, J. Influence of ripeness and maceration of the grapes on levels of furan and carbonyl compounds in wine—Simultaneous quantitative determination and assessment of the exposure risk to these compounds. Food Chem. 2017, 230, 594–603. [Google Scholar] [CrossRef]
- Kächele, M.; Monakhova, Y.B.; Kuballa, T.; Lachenmeier, D.W. NMR investigation of acrolein stability in hydroalcoholic solution as a foundation for the valid HS-SPME/GC–MS quantification of the unsaturated aldehyde in beverages. Anal. Chim. Acta 2014, 820, 112–118. [Google Scholar] [CrossRef]
- Lim, H.H.; Shin, H.S. Simple determination of acrolein in surface and drinking water by headspace SPME GC–MS. Chromatographia 2012, 75, 943–948. [Google Scholar] [CrossRef]
- Ledauphin, J.; Lefrancois, A.; Marquet, N.; Beljean-Leymarie, M.; Barillier, D. Development of an accurate and sensitive gas chromatographic method for the determination of acrolein content in Calvados and cider. LWT Food Sci. Technol. 2006, 39, 1045–1052. [Google Scholar] [CrossRef]
- Wardencki, W.; Sowiński, P.; Curyło, J. Evaluation of headspace solid-phase microextraction for the analysis of volatile carbonyl compounds in spirits and alcoholic beverages. J. Chromatogr. A 2003, 984, 89–96. [Google Scholar] [CrossRef]
- Masson, J.; Cardoso, M.G.; Zacaroni, L.M.; dos Anjos, J.P.; Sackz, A.A.; Machado, A.M.R.; Nelson, D.L. Determination of acrolein, ethanol, volatile acidity, and copper in different samples of sugarcane spirits. Ciênc. Tecnol. Aliment. 2012, 32, 568–572. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, R.F.; Marques, J.C.; Neto, B.S.L.; De Keukeleire, D.; Franco, D.W. Qualitative and quantitative high-performance liquid chromatographic analysis of aldehydes in Brazilian sugar cane spirits and other distilled alcoholic beverages. J. Chromatogr. A 1997, 782, 13–23. [Google Scholar] [CrossRef]
- Lehtonen, P.; Laakso, R.; Puputti, E. Liquid chromatographic determination of 2-propenal (acrolein) and 2-butenal (crotonaldehyde) from water-ethanol mixtures. Z. Lebensm. Unters. Forsch. 1984, 178, 487–489. [Google Scholar] [CrossRef]
- Curyło, J.; Wardencki, W. Determination of acetaldehyde and acrolein in raw spirits by capillary isotachophoresis after derivatization. Anal. Lett. 2005, 38, 1659–1669. [Google Scholar] [CrossRef]
- Baños, C.E.; Silva, M. Analysis of low-molecular mass aldehydes in drinking waters through capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis 2010, 31, 2028–2036. [Google Scholar] [CrossRef]
- Duangdee, N.; Chamboonchu, N.; Kongkiatpaiboon, S.; Prateeptongkum, S. Quantitative 1H NMR spectroscopy for the determination of oxyresveratrol in Artocarpus lacucha heartwood. Phytochem. Anal. 2019, 30, 617–622. [Google Scholar] [CrossRef]
- Dong, J.W.; Cai, L.; Fang, Y.S.; Duan, W.H.; Li, Z.J.; Ding, Z.T. Simultaneous, simple and rapid determination of five bioactive free anthraquinones in Radix et Rhizoma Rhei by quantitative 1H NMR. J. Braz. Chem. Soc. 2016, 27, 2120–2126. [Google Scholar]
- Košir, I.J.; Kidrič, J. Use of modern nuclear magnetic resonance spectroscopy in wine analysis: Determination of minor compounds. Anal. Chim. Acta 2002, 458, 77–84. [Google Scholar] [CrossRef]
- del Campo, G.; Berregi, I.; Caracena, R.; Zuriarrain, J. Quantitative determination of caffeine, formic acid, trigonelline and 5-(hydroxymethyl)furfural in soluble coffees by 1H NMR spectrometry. Talanta 2010, 81, 367–371. [Google Scholar] [CrossRef]
- Liu, M.; Mao, X.; Ye, C.; Huang, H.; Nicholson, J.K.; Lindon, J.C. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J. Magn. Reson. 1998, 132, 125–129. [Google Scholar] [CrossRef]
- Mestrelab Research. Chemistry Software Solutions. Available online: https://mestrelab.com (accessed on 26 November 2020).
- Vold, R.L.; Waugh, J.S. Measurement of spin relaxation in complex systems. J. Chem. Phys. 1968, 48, 3831–3832. [Google Scholar] [CrossRef] [Green Version]
- Berregi, I.; del Campo, G.; Caracena, R.; Miranda, J.I. Quantitative determination of formic acid in apple juices by 1H NMR spectrometry. Talanta 2007, 72, 1049–1053. [Google Scholar] [CrossRef]
- Mahanta, B.P.; Sut, D.; Kemprai, P.; Paw, M.; Lal, M.; Haldar, S. A 1H-NMR spectroscopic method for the analysis of thermolabile chemical markers from the essential oil of black turmeric (Curcuma caesia) rhizome: Application in post-harvest analysis. Phytochem. Anal. 2020, 31, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Li, F.F.; Liu, H.X.; Zhang, Y.L.; Wang, S.Q. 1H NMR determination of 1,3-dicyclohexylurea, glutaric acid and triethylamine in medical four-arm poly(ethylene glycol)-N-hydroxysuccinimideglutarate for better quality control. Anal. Methods 2019, 11, 6176–6183. [Google Scholar] [CrossRef]
- Fernández-Pastor, I.; Luque-Muñoz, A.; Rivas, F.; Medina-O’Donnell, M.; Martinez, A.; González-Maldonado, R.; Haidour, A.; Parra, A. Quantitative NMR analysis of L-Dopa in seeds from two varieties of Mucuna pruriens. Phytochem. Anal. 2019, 30, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, T.; Alves, D.; Santana, C.; Romão, W.; Roberto, P.; Bezerra, R.; de Souza, W.; Conti, R.; Lacerda, V., Jr.; Cunha, A. Quantification of capsaicinoids from chili peppers using 1H NMR without deuterated solvent. Anal. Methods 2019, 11, 1939–1950. [Google Scholar]
- Yu, C.; Zhang, Q.; Xu, P.Y.; Bai, Y.; Shen, W.B.; Di, B.; Su, M.X. Quantitative determination and validation of octreotide acetate using 1H-NMR spectroscopy with internal standard method. Magn. Reson. Chem. 2018, 56, 37–45. [Google Scholar] [CrossRef]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Pearson Education Limited: Harlow, UK, 2010; Chapters 3 and 5. [Google Scholar]
- Zuriarrain, A.; Zuriarrain, J.; Villar, M.; Berregi, I. Quantitative determination of ethanol in cider by 1H NMR spectrometry. Food Control 2015, 50, 758–762. [Google Scholar] [CrossRef]
- del Campo, G.; Berregi, I.; Caracena, R.; Santos, J.I. Quantitative analysis of malic and citric acids in fruit juices using proton nuclear magnetic resonance spectroscopy. Anal. Chim. Acta 2006, 556, 462–468. [Google Scholar] [CrossRef]
- Zuriarrain, A.; Zuriarrain, J.; Puertas, A.I.; Dueñas, M.T.; Berregi, I. Quantitative determination of lactic and acetic acids in cider by 1H NMR spectrometry. Food Control 2015, 52, 49–53. [Google Scholar] [CrossRef]
- Berregi, I.; Santos, J.I.; del Campo, G.; Miranda, J.I.; Aizpurua, J.M. Quantitative determination of chlorogenic acid in cider apple juices by 1H NMR spectrometry. Anal. Chim. Acta 2003, 486, 269–274. [Google Scholar] [CrossRef]
- Berregi, I.; Santos, J.I.; del Campo, G.; Miranda, J.I. Quantitative determination of (−)-epicatechin in cider apple juices by 1H NMR. Talanta 2003, 61, 139–145. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cider | * Concentration, mg/L | |
|---|---|---|
| 1H NMR | GC | |
| 1 | 9.8 ± 0.2 | 10.3 ± 0.3 |
| 2 | 6.0 ± 0.1 | 5.7 ± 0.2 |
| 3 | 2.6 ± 0.1 | 2.5 ± 0.1 |
| 4 | 6.8 ± 0.2 | 6.7 ± 0.2 |
| 5 | 2.3 ± 0.1 | 2.4 ± 0.1 |
| 6 | 12.3 ± 0.3 | 12.8 ± 0.4 |
| 7 | 31.8 ± 0.7 | 32.5 ± 0.9 |
| 8 | 15.6 ± 0.4 | 15.2 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de las Heras, E.; Zuriarrain-Ocio, A.; Zuriarrain, J.; Bordagaray, A.; Dueñas, M.T.; Berregi, I. Quantitative Determination of Acrolein in Cider by 1H NMR Spectrometry. Foods 2020, 9, 1820. https://doi.org/10.3390/foods9121820
de las Heras E, Zuriarrain-Ocio A, Zuriarrain J, Bordagaray A, Dueñas MT, Berregi I. Quantitative Determination of Acrolein in Cider by 1H NMR Spectrometry. Foods. 2020; 9(12):1820. https://doi.org/10.3390/foods9121820
Chicago/Turabian Stylede las Heras, Enaitz, Andoni Zuriarrain-Ocio, Juan Zuriarrain, Ane Bordagaray, María Teresa Dueñas, and Iñaki Berregi. 2020. "Quantitative Determination of Acrolein in Cider by 1H NMR Spectrometry" Foods 9, no. 12: 1820. https://doi.org/10.3390/foods9121820
APA Stylede las Heras, E., Zuriarrain-Ocio, A., Zuriarrain, J., Bordagaray, A., Dueñas, M. T., & Berregi, I. (2020). Quantitative Determination of Acrolein in Cider by 1H NMR Spectrometry. Foods, 9(12), 1820. https://doi.org/10.3390/foods9121820