1. Introduction
Paprika is a red spice obtained after drying and grinding some varieties of peppers of the genus
Capsicum belonging to the large family of Solanaceae [
1]. The five main cultivated and economically important species of paprika are
Capsicum annuum L.,
C. chinense Jacq. and
C. frutescens L., now widely grown throughout Europe, the southern United States, Africa, India and China, and the species
C. baccatum L. and
C. pubescens Ruiz & Pav., which are grown predominantly in South America [
2,
3].
Paprika is often used in many foods such as soup, meat, ice cream, baked goods, and in seasoning blends to add color and taste [
4], although it is also used in personal protection sprays, medicine, and cosmetics [
5,
6,
7,
8,
9].
Today, both consumers and food manufacturers are increasingly concerned about quality standards and as a result, there is a growing demand for food traceability. In this context, the designation of Protected Designation of Origin (PDO) is an effective tool to guarantee the quality and geographical origin of a product. In Europe, there are five Protected Designations of Origin for paprika: Piment d’Espelette (France), Paprika Szeged (Hungary), Paprika Žitava (Slovakia Žitava paprika), Pimentón de La Vera (Cáceres, Extremadura, Spain), and Pimentón de Murcia (Murcia, Spain).
To protect these products, the competent authorities carry out various checks, consisting mainly of inspections of production sites. During these inspections, records, raw materials, production systems, maturation, etc. are checked and various samples are taken to be analyzed by independent laboratories to ensure that the whole process complies with the regulations in force.
However, adulteration in natural products remains a widespread practice that mainly seeks economic benefit, either through increased sales or through reduced production costs. This practice is very difficult to combat, mainly due to the globalization of trade, the complexity of supply chains, the regulatory policy differences between each country and the fact that the main legal responsibility for the safety of marketed products is delegated by default to manufacturers [
10]. Besides, the methods used for the adulteration of natural products are increasingly sophisticated, and, in some cases, are specially designed to mislead the methods of analysis applied by the competent authorities, which are limited to routine analyses that do not detect adulterations.
However, the high number of illegal compounds, as well as the different ways of adulterating a natural product, makes it sometimes difficult to identify those compounds that should be analyzed. In this context, non-targeted methods encompass the complexity of modern authentication of natural products [
11,
12,
13,
14]. The main focus of this type of strategy is to detect as many compounds as possible below 1.500 Da [
15]. For this reason, liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS), such as TOF and Orbitrap, as well as their hybrid configurations, are the most suitable instruments to carry out this type of strategy, since they present a high sensitivity, have a high-resolution power (over 100,000 FWHM, full width at half maximum) and a very accurate mass/charge ratio determination (<5 ppm), able to distinguish between isobaric compounds. Furthermore, data acquisition in high-resolution full scan mode allows the simultaneous combination of targeted and non-targeted analysis, identification of new compounds, and retrospective data analysis [
16]. Another important feature of this analytical strategy is that the authentication of a product can be approached from various perspectives in a single analysis. Depending on the selected approach to solve the problem of interest, the fingerprint allows not only detecting the origin or type of raw material used but also detects unlabeled compounds, unauthorized additives or the use of prohibited technological processes, among others [
17].
In this type of analytical strategy, sample preparation must be as simple and non-specific as possible, to avoid losses of compounds, accompanied by loss of information [
17]. Normally, the sample is extracted with a hydrophilic organic solvent-water mixture.
In non-targeted methods based on fingerprinting, it is not necessary to identify the compounds that are most relevant to product authentication. This type of strategy is often qualitative and based on a comparison between the samples under study and the authentic reference samples, which are used to build an appropriate database. This database is used to compare the fingerprints of the unknown samples with the fingerprints of the authentic reference samples, thus reducing the time and cost of analysis [
12]. However, identification is important when regulatory action must be taken based on the results. In a court of law, the identification of the chemical structure of a primary or secondary marker is an important asset in the judicial process [
11], although it should be kept in mind that different biomarkers can be obtained depending on the procedure used. The biomarkers obtained depend largely on the overall experimental approach used, including sample handling, separation, and detection, as well as the specific instrumentation used [
18].
Analytical signals can be obtained in positive mode, in negative mode or by data fusion. In many cases, the most important aspect is to detect as many signals as possible, under both positive and negative modes and joining all the information in a single matrix, that is by data fusion, a complex authentication problem could be achieved. Another important feature of HRMS instruments such as q-Orbitrap, is their ability to automatically isolate those signals that are more abundant, to be fragmented for future identification, if deemed appropriate.
Data pre-processing is one of the most relevant points to consider in methods based on fingerprinting. Once the data has been generated, it is important to have software that helps to obtain the working matrix. This matrix is constituted by the retention time, the
m/z values and the area or signal of each detected peak. In this software, some parameters must be set, such as the mass tolerance for peak alignment, the total intensity threshold, the maximum peak displacement, and the S/N threshold. Sometimes, these parameters eliminate chemical interferences from the matrix, but they must be properly adjusted to prevent discarding signals that may be relevant [
18]. Besides, depending on the type of study being conducted, it is necessary to reduce the number of signals to simplify the matrix obtained. In these cases, some actions can be taken such as removing signals that are not detected in a minimum percentage of the samples or remove signals that are not observed in the quality controls, which generally consist of a mixture of equal volumes of all the samples analyzed [
18].
Once the data matrix has been obtained, it should be evaluated using multivariate statistical models, whether supervised or unsupervised, that finally allow the authentication of the product under study [
13]. However, the great diversity of chemometric models, together with the fact that this type of strategy is in its beginnings in the food field, make that there is not yet a clear consensus as to which chemometric model is most suitable for routine use in quality control.
The main objective of this work was to develop a UHPLC-HRMS (Orbitrap) method for the characterization, classification, and authentication of paprika samples using a non-targeted fingerprint approach. Different samples of La Vera paprika, Murcia paprika, and Czech Republic paprika were analyzed using a simple sample extraction procedure. The hypothesis established in this work is that UHPLC-HRMS fingerprint data, obtained in ESI negative mode, can be considered as a source of potential chemical descriptors to be exploited for the characterization and classification of paprika samples by unsupervised and supervised methods such as principal components analysis (PCA) and partial least squares regression-discriminant analysis (PLS-DA).
2. Materials and Methods
2.1. Chemicals and Standard Solutions
All the reagents, standards and chemicals employed in the present work were of analytical grade. Water, acetonitrile, and methanol (all of them LC-MS Chromasolv® quality), acetone, and formic acid (98–100%) were purchased from Sigma-Aldrich (Steinheim, Germany). Hydrochloric acid (35%) was obtained from Merck (Seelze, Germany).
2.2. Instrumentation
An Accela UHPLC instrument from Thermo Fisher Scientific (San Jose, CA, USA), with a quaternary pump, and an autosampler, was employed for the sample chromatographic analysis. Reversed-phase separation in an Ascentis® Express C18 porous-shell (150 × 2.1 mm, 2.7 µm partially porous particle size) column obtained from Supelco (Bellefonte, PA, USA) under universal gradient elution mode using water (solvent A) and acetonitrile (solvent B), both of them containing 0.1% formic acid, was proposed for obtaining the chromatographic fingerprints. The elution gradient program employed begun with an isocratic elution step at 10% B for 1 min, followed by a linear gradient from 10 to 95% solvent B in 19 min. Then, 95% solvent B was kept for 3 min, and back to initial conditions at 10% solvent B in 1 min. A column re-equilibration time of 6 min at 10% solvent B was employed, giving place to a total chromatographic gradient program of 30 min. The mobile phase flow rate was 300 µL/min. The column was kept at room temperature, and an injection volume of 10 µL (at full-loop mode) was employed for sample analysis.
The UHPLC instrument was coupled to a Q-Exactive Orbitrap HRMS instrument (Thermo Fisher Scientific) by employing a heated electrospray ionization source (H-ESI II). Nitrogen (purity of 99.98%) was employed for the H-ESI sheath, sweep, and auxiliary gases at flow rates of 60, 0, and 10 a.u. (arbitrary units). H-ESI was operated in negative ionization mode by applying a capillary voltage of −2.5 kV. H-ESI vaporizer temperature and capillary instrument temperature were kept at 350 °C and 320 °C, respectively. An S-Lens RF level of 50 V was used. Tuning and calibration of the Orbitrap analyzer were performed every 3 days by using the Thermo Fisher Scientific commercially available calibration solution for that purpose. Full scan HRMS spectra (m/z 100–1500) at a mass resolution of 70.000 FWHM (full-width at half maximum, at m/z 200) was employed to register the UHPLC-HRMS metabolomics fingerprints, with an automatic gain control (AGC) target (which is the number of ions to fill the instrument C-Trap) of 2.5 × 105, and a maximum injection time of 200 ms.
UHPLC-HRMS system control and data processing were performed using Xcalibur version 3.1 software (Thermo Fisher Scientific).
2.3. Samples and Sample Treatment
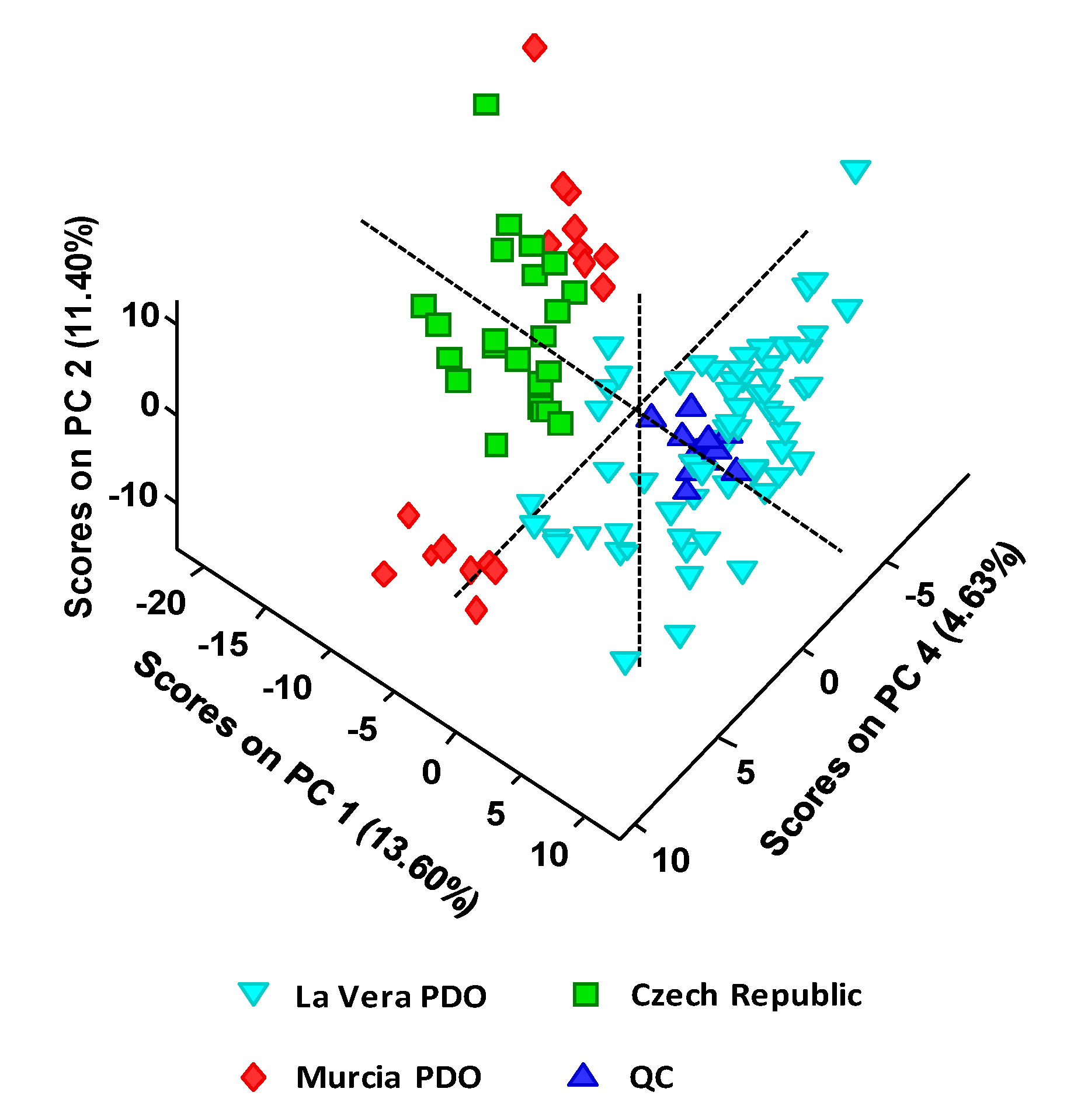
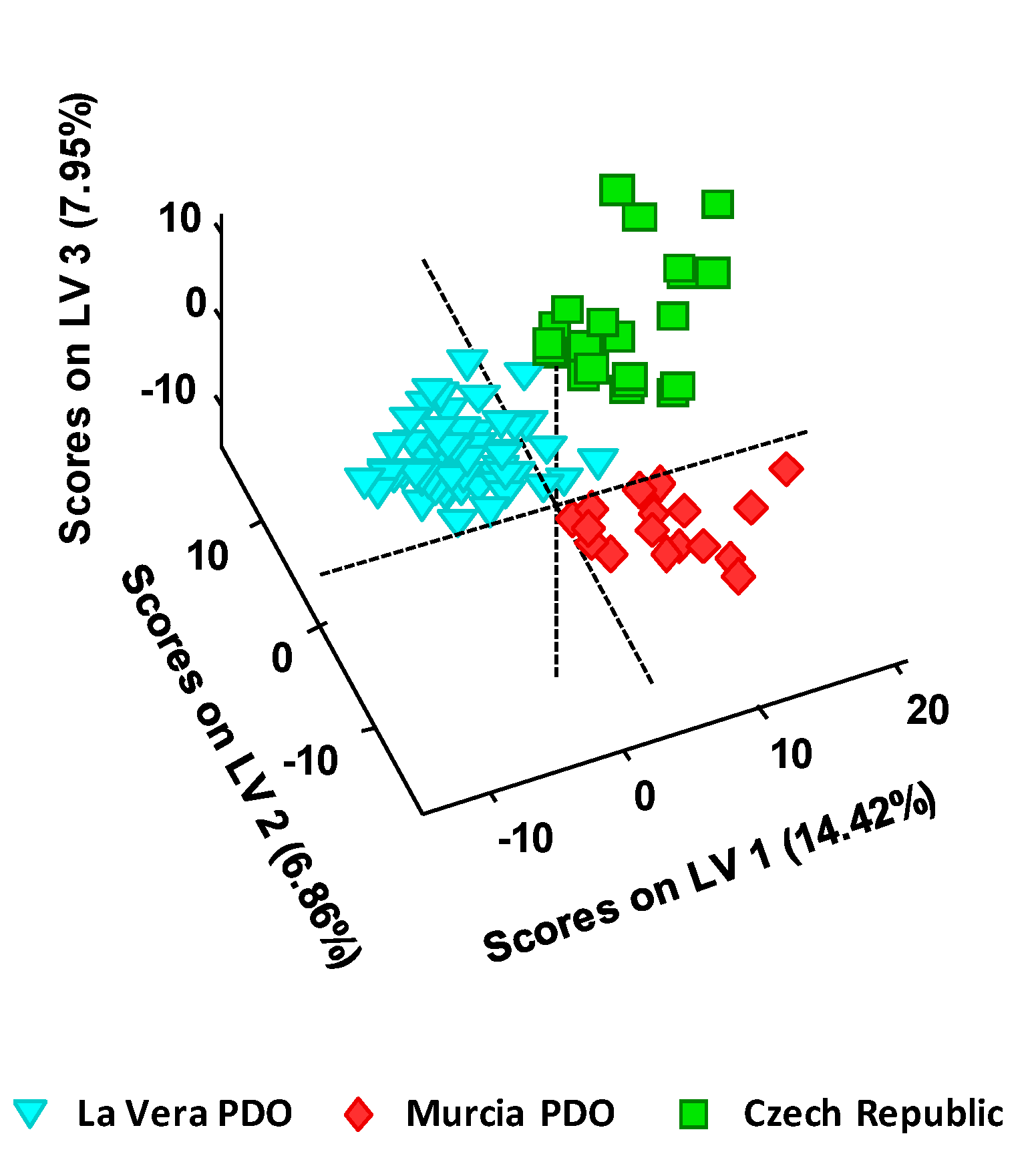
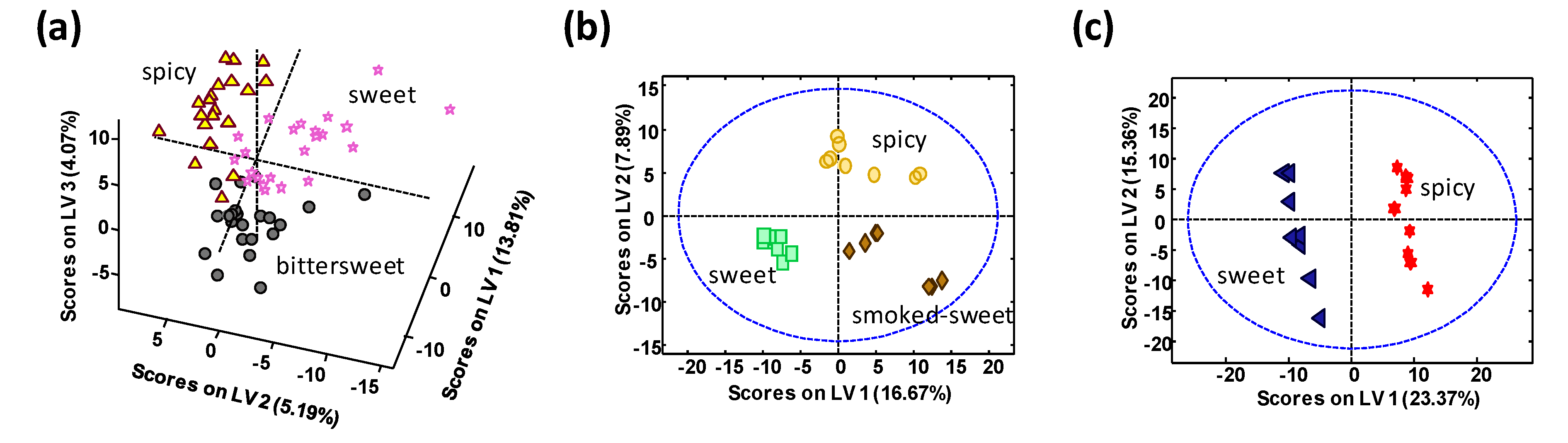
One hundred and five paprika samples obtained from local markets in Spain and the Czech Republic were analyzed. Samples belong to different PDO and production regions, as well as different taste varieties: 65 samples from La Vera PDO (including 23 sweet, 22 bittersweet, and 20 spicy), 17 samples from Murcia PDO (including 8 sweet, and 9 spicy), and 23 samples from Czech Republic (including 7 smoked-sweet, 8 sweet, and 8 spicy).
Samples were extracted following a previously proposed procedure [
1,
19]. Briefly, paprika samples (0.3 mg) were extracted with water:acetonitrile 20:80
v/v solution (3 mL) by stirring (1 min) with a vortex mixer (Stuart, Sone, UK), and by sonication (15 min) with an ultrasonic bath (2510 Branson, Hampton, NH, USA). Centrifugation was then carried out for 15 min at 4500 rpm (Rotana 460 HR centrifuge, Hettich, Germany). The obtained extract was then filtered with 0.45 µm nylon filters (Whatman, Clifton, NJ, USA) and transferred into 2 mL injection vials, which were kept at −18 °C until the UHPLC-HRMS analysis.
Besides, a quality control (QC) solution, employed for the evaluation of the method reproducibility and to ensure the robustness of the chemometric results, was employed. This QC was prepared by mixing 50 µL of each one of the paprika sample extracts obtained.
To prevent signal tendencies attributed to the sample sequence analysis, all paprika samples were analyzed randomly with the proposed UHPLC-HRMS method. Besides, blanks of acetonitrile and QCs were injected every 10 randomly analyzed samples (representing 12 QC analyses).
2.4. Data Analysis
Data matrices for untargeted UHPLC-HRMS analysis were obtained with R software (R Foundation, Vienna, Austria). First, UHPLC-HRMS raw data was submitted to MSConvert software to obtain an Excel file with the profile of peak intensities as a function of
m/z values and retention times for all the chemical features detected. An absolute intensity threshold peak filter of 10
5 was employed. PCA and PLS-DA chemometric calculations were made by using SOLO 8.6 chemometric software (Eigenvector Research [
20], Manson, WA, USA). The theoretical background of these methods in a detailed way is addressed elsewhere [
21]. X-data matrices for PCA and PLS-DA were based on the UHPLC-HRMS metabolomic fingerprints (peak intensities as a function of retention time and
m/
z values) obtained in H-ESI (-) mode. The PLS-DA Y-data matrix included the analyzed sample classes. Scatter plots of scores of the principal components (PCs) in PCA and the latent variables (LVs) in PLS-DA were used to study the distribution and classification of samples. The applicability of the proposed PLS-DA models for sample classification was assessed by employing 70% of the analyzed samples as the calibration sets (71 samples), while the remaining 30% of samples were used for prediction and validation (31 samples). Optimal number of LV in PLS-DA was determined by considering the first significant minimum point of the cross-validation (CV) error from a Venetian blind approach.
4. Conclusions
In the present work, the feasibility of non-targeted UHPLC-HRMS (Orbitrap) fingerprints as appropriate sample chemical descriptors for the characterization, classification, and authentication of paprika samples according to both their PDO and production region and their different taste varieties has been demonstrated.
The proposed characterization and classification method has the advantage that the identification of specific metabolite compounds is not required to deal with sample authentication, as non-targeted fingerprints based on HRMS signal intensities as a function of m/z values and retention times are treated by the chemometric approaches.
Unsupervised exploratory analysis performed by PCA and supervised classification carried out by PLS-DA by using the obtained non-targeted UHPLC-HRMS fingerprints showed excellent discrimination capabilities of the different paprika production regions under study (La Vera PDO, Murcia PDO, and the Czech Republic paprika). PLS-DA model validations resulted in 100% classification rates in both calibration and prediction steps.
Besides, the proposed methodology also exhibited perfect discrimination and authentication capabilities among the different paprika taste varieties of each of the production regions studied—even in the case of La Vera PDO samples, where three different taste varieties (sweet, bittersweet, and spicy) are produced within a small geographical area in comparison to Murcia PDO and the Czech Republic samples.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}