
Developmental Programming: Impact of Prenatal Exposure to Bisphenol A on Senescence and Circadian Mediators in the Liver of Sheep



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
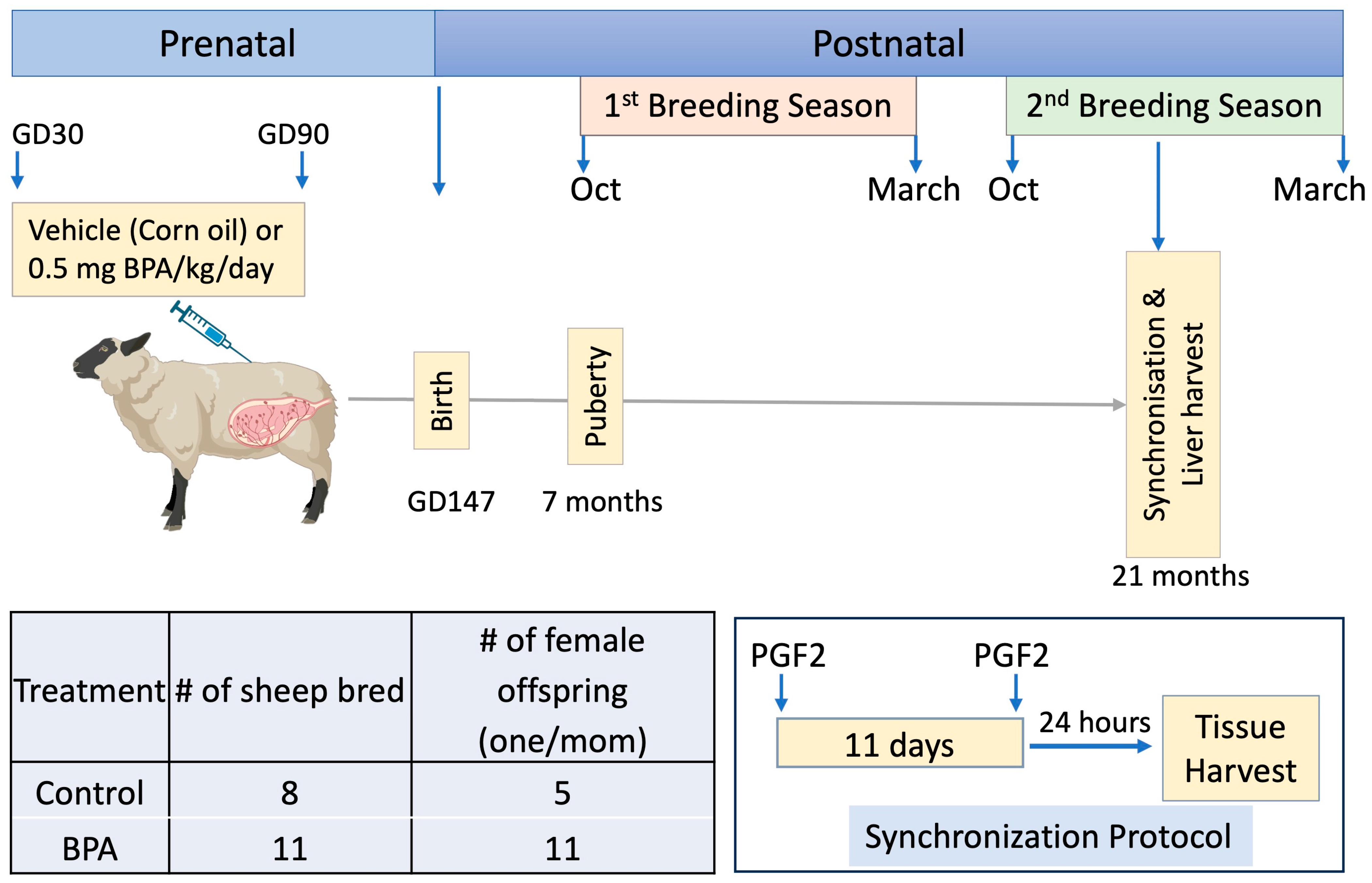
2.1. Animals
2.2. Prenatal BPA Treatments
2.3. Tissue Collection
2.4. RT-PCR
2.4.1. mRNA Expression of Circadian Genes and Aging Genes
2.4.2. Mitochondrial DNA Copy Number
2.4.3. Telomere Length Assay
2.5. Lipofuscin Staining
2.6. Statistical Analysis
3. Results
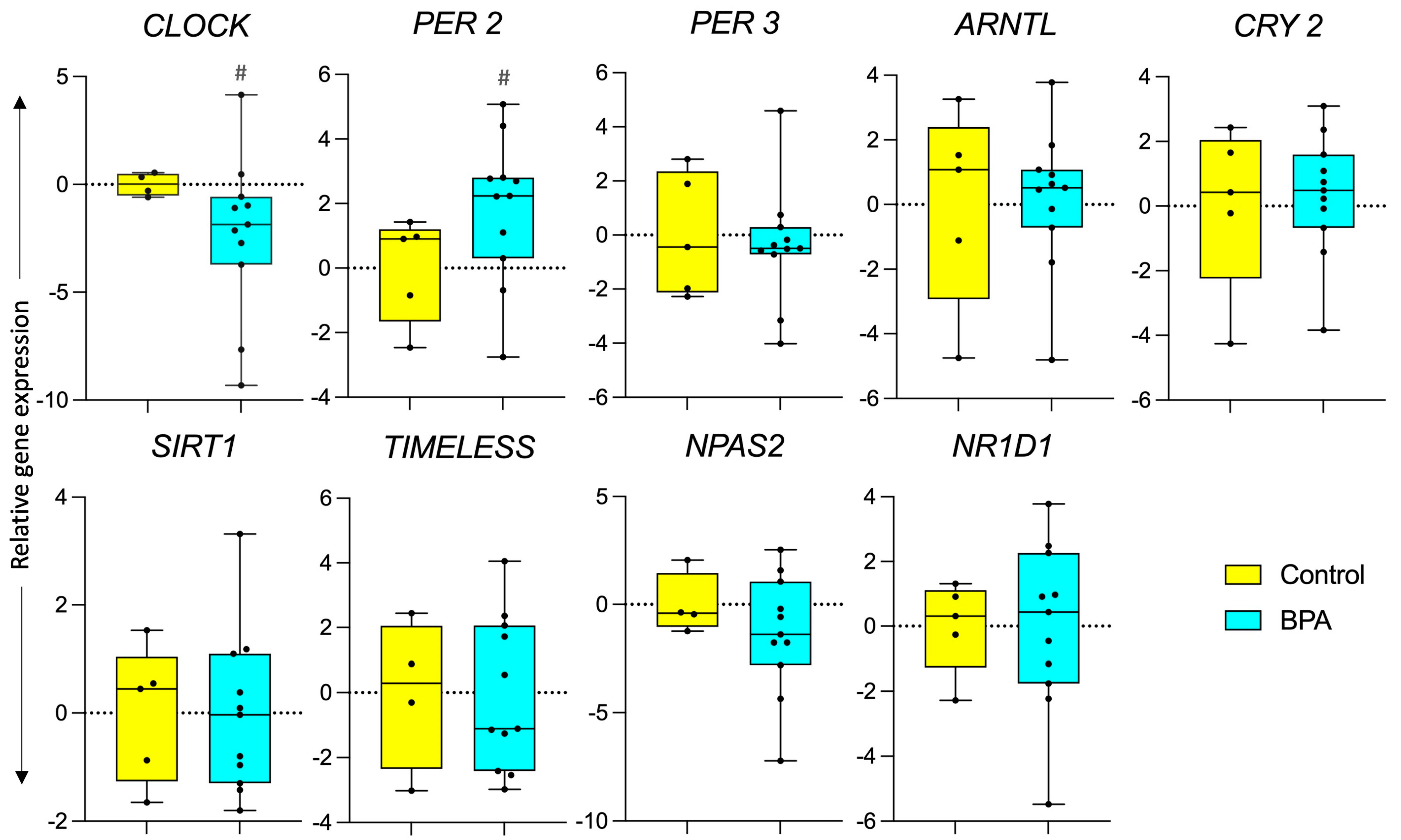
3.1. Effect of Prenatal BPA Exposure on Circadian Function
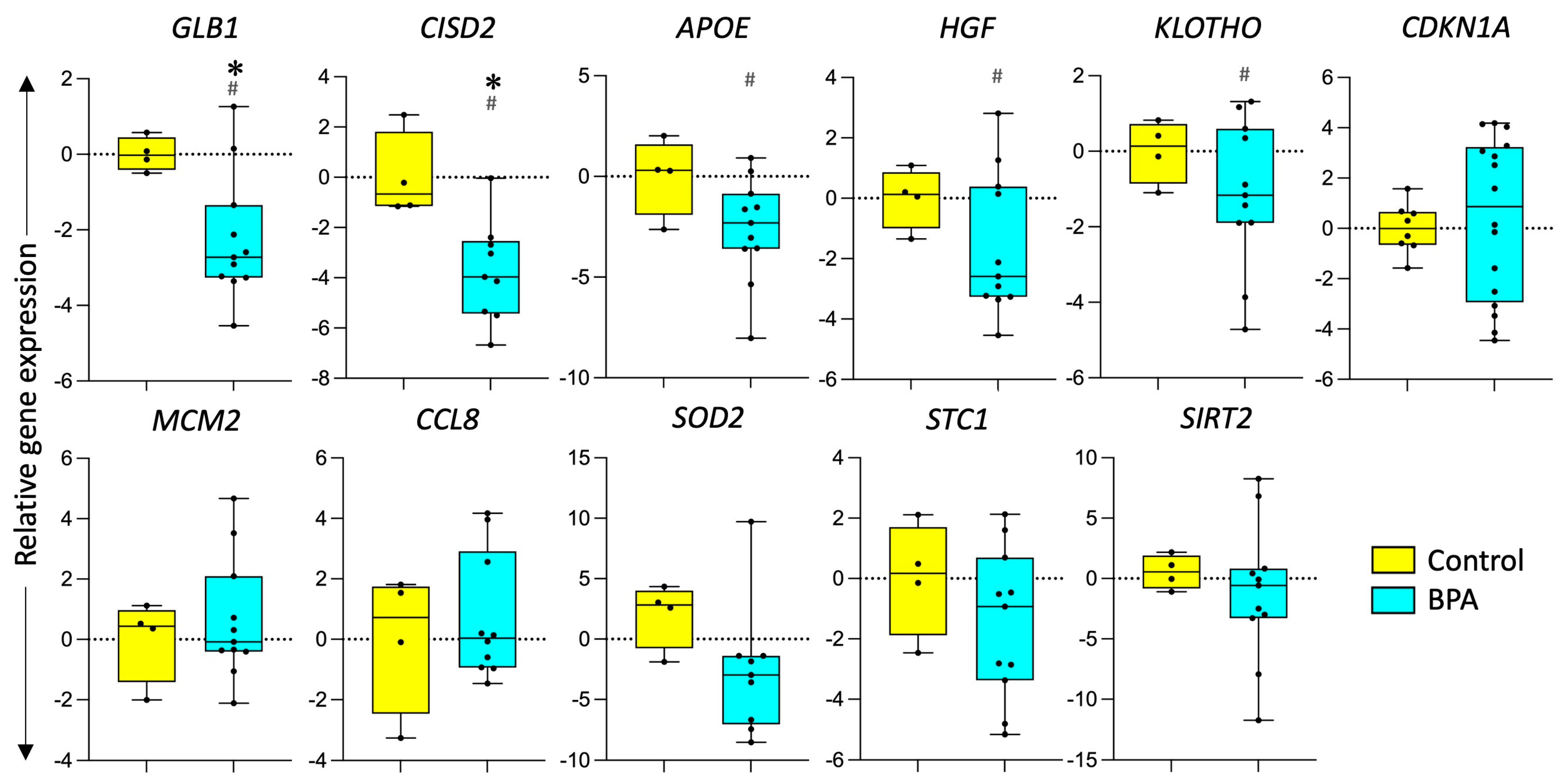
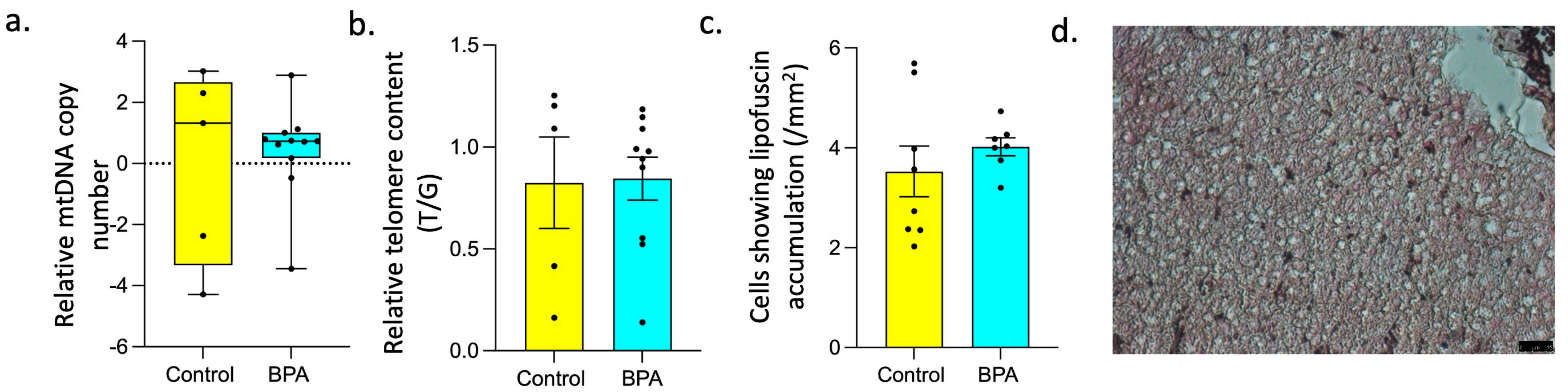
3.2. Effect of Prenatal BPA Exposure on Markers of Longevity and Senescence
4. Discussion
4.1. Impact of Prenatal BPA on Circadian Genes
4.2. Impact of Prenatal BPA on Longevity and Senescence Genes
4.3. Impact of Prenatal BPA on Other Markers of Senescence
4.4. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Le, M.H.; Yeo, Y.H.; Zou, B.; Barnet, S.; Henry, L.; Cheung, R.; Nguyen, M.H. Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical bayesian approach. Clin. Mol. Hepatol. 2022, 28, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Powell, T.L.; Barrett, E.S.; Hardy, D.B. Developmental origins of metabolic diseases. Physiol. Rev. 2021, 101, 739–795. [Google Scholar] [CrossRef]
- de Gusmao Correia, M.L.; Volpato, A.M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Developmental origins of health and disease: Experimental and human evidence of fetal programming for metabolic syndrome. J. Hum. Hypertens. 2012, 26, 405–419. [Google Scholar] [CrossRef]
- Heindel, J.J. The developmental basis of disease: Update on environmental exposures and animal models. Basic. Clin. Pharmacol. Toxicol. 2019, 125 (Suppl. 3), 5–13. [Google Scholar] [CrossRef]
- Hsu, C.N.; Hou, C.Y.; Hsu, W.H.; Tain, Y.L. Early-Life Origins of Metabolic Syndrome: Mechanisms and Preventive Aspects. Int. J. Mol. Sci. 2021, 22, 11872. [Google Scholar] [CrossRef]
- Galvan-Martinez, D.H.; Bosquez-Mendoza, V.M.; Ruiz-Noa, Y.; Ibarra-Reynoso, L.D.R.; Barbosa-Sabanero, G.; Lazo-de-la-Vega-Monroy, M.L. Nutritional, pharmacological, and environmental programming of NAFLD in early life. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G99–G114. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Song, W.; Puttabyatappa, M. Praegnatio Perturbatio-Impact of Endocrine-Disrupting Chemicals. Endocr. Rev. 2021, 42, 295–353. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, G.; Bhaskaran, R.S.; Karundevi, B. Developmental exposure to DEHP alters hepatic glucose uptake and transcriptional regulation of GLUT2 in rat male offspring. Toxicology 2019, 413, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Mao, Y.; Wang, J.; Yu, H.; Zhang, X.; Pei, X.; Duan, Z.; Xiao, C.; Ma, M. Gestational bisphenol A exposure impairs hepatic lipid metabolism by altering mTOR/CRTC2/SREBP1 in male rat offspring. Hum. Exp. Toxicol. 2022, 41, 9603271221129852. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Chakraborty, P. A review on sources and health impacts of bisphenol A. Rev. Environ. Health 2020, 35, 201–210. [Google Scholar] [CrossRef]
- Gerona, R.R.; Pan, J.; Zota, A.R.; Schwartz, J.M.; Friesen, M.; Taylor, J.A.; Hunt, P.A.; Woodruff, T.J. Direct measurement of Bisphenol A (BPA), BPA glucuronide and BPA sulfate in a diverse and low-income population of pregnant women reveals high exposure, with potential implications for previous exposure estimates: A cross-sectional study. Environ. Health 2016, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Zbucka-Kretowska, M.; Lazarek, U.; Miltyk, W.; Sidorkiewicz, I.; Pierzynski, P.; Milewski, R.; Wolczynski, S.; Czerniecki, J. Simultaneous analysis of bisphenol A fractions in maternal and fetal compartments in early second trimester of pregnancy. J. Perinat. Med. 2019, 47, 765–770. [Google Scholar] [CrossRef]
- Nahar, M.S.; Liao, C.; Kannan, K.; Dolinoy, D.C. Fetal liver bisphenol A concentrations and biotransformation gene expression reveal variable exposure and altered capacity for metabolism in humans. J. Biochem. Mol. Toxicol. 2013, 27, 116–123. [Google Scholar] [CrossRef]
- Vom Saal, F.S.; Vandenberg, L.N. Update on the Health Effects of Bisphenol A: Overwhelming Evidence of Harm. Endocrinology 2021, 162, bqaa171. [Google Scholar] [CrossRef]
- Manzan-Martins, C.; Paulesu, L. Impact of bisphenol A (BPA) on cells and tissues at the human materno-fetal interface. Tissue Cell 2021, 73, 101662. [Google Scholar] [CrossRef]
- Nahar, M.S.; Kim, J.H.; Sartor, M.A.; Dolinoy, D.C. Bisphenol A-associated alterations in the expression and epigenetic regulation of genes encoding xenobiotic metabolizing enzymes in human fetal liver. Environ. Mol. Mutagen. 2014, 55, 184–195. [Google Scholar] [CrossRef]
- Abulehia, H.F.S.; Mohd Nor, N.S.; Sheikh Abdul Kadir, S.H. The Current Findings on the Impact of Prenatal BPA Exposure on Metabolic Parameters: In Vivo and Epidemiological Evidence. Nutrients 2022, 14, 2766. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Prenatal Exposure to BPA and Offspring Outcomes: The Diabesogenic Behavior of BPA. Dose-Response 2015, 13, 1559325815590395. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, F.; Aquilina, A.; Vassallo, J.; Pace, N.P. Bisphenol A and Type 2 Diabetes Mellitus: A Review of Epidemiologic, Functional, and Early Life Factors. Int. J. Environ. Res. Public Health 2021, 18, 716. [Google Scholar] [CrossRef] [PubMed]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Dabeer, S.; Raisuddin, S. Perinatal exposure to environmental endocrine disruptor bisphenol A aggravates the onset of non-alcoholic fatty liver disease (NAFLD) in weanling F1 offspring of obese rats. Environ. Sci. Pollut. Res. Int. 2023, 30, 3146–3165. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Fan, J.; Wu, G.; Liu, X.; Wu, H.; Liu, J.; Chen, Y.; Su, S.; Cheng, X.; Xu, Z.; et al. Gestational bisphenol A exposure induces fatty liver development in male offspring mice through the inhibition of HNF1b and upregulation of PPARgamma. Cell Biol. Toxicol. 2021, 37, 65–84. [Google Scholar] [CrossRef]
- Banstola, A.; Reynolds, J.N.J. The Sheep as a Large Animal Model for the Investigation and Treatment of Human Disorders. Biology 2022, 11, 1251. [Google Scholar] [CrossRef]
- Enkhbaatar, P.; Nelson, C.; Salsbury, J.R.; Carmical, J.R.; Torres, K.E.; Herndon, D.; Prough, D.S.; Luan, L.; Sherwood, E.R. Comparison of Gene Expression by Sheep and Human Blood Stimulated with the TLR4 Agonists Lipopolysaccharide and Monophosphoryl Lipid A. PLoS ONE 2015, 10, e0144345. [Google Scholar] [CrossRef]
- Murray, S.J.; Mitchell, N.L. The Translational Benefits of Sheep as Large Animal Models of Human Neurological Disorders. Front. Vet. Sci. 2022, 9, 831838. [Google Scholar] [CrossRef]
- Padmanabhan, V.; Veiga-Lopez, A. Sheep models of polycystic ovary syndrome phenotype. Mol. Cell. Endocrinol. 2013, 373, 8–20. [Google Scholar] [CrossRef]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Pilz, P.M.; Ward, J.E.; Chang, W.T.; Kiss, A.; Bateh, E.; Jha, A.; Fisch, S.; Podesser, B.K.; Liao, R. Large and Small Animal Models of Heart Failure With Reduced Ejection Fraction. Circ. Res. 2022, 130, 1888–1905. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Veiga-Lopez, A. Animal models of the polycystic ovary syndrome phenotype. Steroids 2013, 78, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Veiga-Lopez, A. Reproduction Symposium: Developmental programming of reproductive and metabolic health. J. Anim. Sci. 2014, 92, 3199–3210. [Google Scholar] [CrossRef] [PubMed]
- Rozance, P.J.; Jones, A.K.; Bourque, S.L.; D’Alessandro, A.; Hay, W.W., Jr.; Brown, L.D.; Wesolowski, S.R. Effects of chronic hyperinsulinemia on metabolic pathways and insulin signaling in the fetal liver. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E721–E733. [Google Scholar] [CrossRef] [PubMed]
- Puttabyatappa, M.; Martin, J.D.; Andriessen, V.; Stevenson, M.; Zeng, L.; Pennathur, S.; Padmanabhan, V. Developmental programming: Changes in mediators of insulin sensitivity in prenatal bisphenol A-treated female sheep. Reprod. Toxicol. 2019, 85, 110–122. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Moeller, J.; Sreedharan, R.; Singer, K.; Lumeng, C.; Ye, W.; Pease, A.; Padmanabhan, V. Developmental programming: Interaction between prenatal BPA exposure and postnatal adiposity on metabolic variables in female sheep. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E238–E247. [Google Scholar] [CrossRef]
- Puttabyatappa, M.; Saadat, N.; Elangovan, V.R.; Dou, J.; Bakulski, K.; Padmanabhan, V. Developmental programming: Impact of prenatal bisphenol-A exposure on liver and muscle transcriptome of female sheep. Toxicol. Appl. Pharmacol. 2022, 451, 116161. [Google Scholar] [CrossRef]
- Tonini, C.; Segatto, M.; Bertoli, S.; Leone, A.; Mazzoli, A.; Cigliano, L.; Barberio, L.; Mandala, M.; Pallottini, V. Prenatal Exposure to BPA: The Effects on Hepatic Lipid Metabolism in Male and Female Rat Fetuses. Nutrients 2021, 13, 1970. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Li, L.; Eguchi, A.; Kannan, K.; Kim, E.Y.; Iwata, H. Effects on the liver lipidome of rat offspring prenatally exposed to bisphenol A. Sci. Total Environ. 2021, 759, 143466. [Google Scholar] [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Shetty, A.; Hsu, J.W.; Manka, P.P.; Syn, W.K. Role of the Circadian Clock in the Metabolic Syndrome and Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2018, 63, 3187–3206. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and liver disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Li, Q.; Li, J. Understanding the Unique Microenvironment in the Aging Liver. Front. Med. 2022, 9, 842024. [Google Scholar] [CrossRef]
- Tahara, Y.; Shibata, S. Circadian rhythms of liver physiology and disease: Experimental and clinical evidence. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 217–226. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Li, L.; Eguchi, A.; Agusa, T.; Yamamoto, K.; Kannan, K.; Kim, E.Y.; Iwata, H. Effects of gestational exposure to bisphenol A on the hepatic transcriptome and lipidome of rat dams: Intergenerational comparison of effects in the offspring. Sci. Total Environ. 2022, 826, 153990. [Google Scholar] [CrossRef]
- Sadria, M.; Layton, A.T. Aging affects circadian clock and metabolism and modulates timing of medication. iScience 2021, 24, 102245. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, X. Aging Disrupts Circadian Rhythms in Mouse Liver Mitochondria. Molecules 2023, 28, 4432. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.S.; Brenner, C.; Masri, S.; Benitah, S.A.; Sassone-Corsi, P. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677.e11. [Google Scholar] [CrossRef] [PubMed]
- National Research Council; Committee for the Update of the Guide for the Care and Use of Laboratory Animals; Institute for Laboratory Animal Research; Division on Earth and Life Studies. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Manikkam, M.; Crespi, E.J.; Doop, D.D.; Herkimer, C.; Lee, J.S.; Yu, S.; Brown, M.B.; Foster, D.L.; Padmanabhan, V. Fetal programming: Prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology 2004, 145, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Reale, E.; Vernez, D.; Hopf, N.B. Skin Absorption of Bisphenol A and Its Alternatives in Thermal Paper. Ann. Work. Expo. Health 2021, 65, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Thayer, K.A.; Taylor, K.W.; Garantziotis, S.; Schurman, S.H.; Kissling, G.E.; Hunt, D.; Herbert, B.; Church, R.; Jankowich, R.; Churchwell, M.I.; et al. Bisphenol A, Bisphenol S, and 4-Hydroxyphenyl 4-Isoprooxyphenyl sulfone (BPSIP) in Urine and Blood of Cashiers. Environ. Health Perspect. 2016, 124, 437–444. [Google Scholar] [CrossRef]
- Loganathan, S.N.; Kannan, K. Occurrence of bisphenol A in indoor dust from two locations in the eastern United States and implications for human exposures. Arch. Environ. Contam. Toxicol. 2011, 61, 68–73. [Google Scholar] [CrossRef]
- Hines, C.J.; Christianson, A.L.; Jackson, M.V.; Ye, X.Y.; Pretty, J.R.; Arnold, J.E.; Calafat, A.M. An Evaluation of the Relationship among Urine, Air, and Hand Measures of Exposure to Bisphenol A (BPA) in US Manufacturing Workers. Ann. Work Expo. Health 2018, 62, 840–851. [Google Scholar] [CrossRef]
- Xue, J.; Wan, Y.; Kannan, K. Occurrence of bisphenols, bisphenol A diglycidyl ethers (BADGEs), and novolac glycidyl ethers (NOGEs) in indoor air from Albany, New York, USA, and its implications for inhalation exposure. Chemosphere 2016, 151, 1–8. [Google Scholar] [CrossRef]
- Taylor, J.A.; Welshons, W.V.; Vom Saal, F.S. No effect of route of exposure (oral; subcutaneous injection) on plasma bisphenol A throughout 24h after administration in neonatal female mice. Reprod. Toxicol. 2008, 25, 169–176. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Luense, L.J.; Christenson, L.K.; Padmanabhan, V. Developmental programming: Gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology 2013, 154, 1873–1884. [Google Scholar] [CrossRef] [PubMed]
- Gerona, R.R.; Woodruff, T.J.; Dickenson, C.A.; Pan, J.; Schwartz, J.M.; Sen, S.; Friesen, M.W.; Fujimoto, V.Y.; Hunt, P.A. Bisphenol-A (BPA), BPA glucuronide, and BPA sulfate in midgestation umbilical cord serum in a northern and central California population. Environ. Sci. Technol. 2013, 47, 12477–12485. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.C.; Driver, H.S. Circadian rhythms, sleep, and the menstrual cycle. Sleep. Med. 2007, 8, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.J.; Sellix, M.T.; Kudo, T.; Nakao, N.; Yoshimura, T.; Ebihara, S.; Colwell, C.S.; Block, G.D. Influence of the estrous cycle on clock gene expression in reproductive tissues: Effects of fluctuating ovarian steroid hormone levels. Steroids 2010, 75, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.X.; Nie, H.T.; Xu, C.J.; Zhang, G.M.; Sun, L.W.; Zhang, T.T.; Wang, Z.; Feng, X.; You, P.H.; Wang, F. Effects of nutrient restriction and arginine treatment on oxidative stress in the ovarian tissue of ewes during the luteal phase. Theriogenology 2018, 113, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P.; Ryde, I.T.; Sanders, L.H.; Howlett, E.H.; Colton, M.D.; Germ, K.E.; Mayer, G.D.; Greenamyre, J.T.; Meyer, J.N. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 2015, 1241, 23–38. [Google Scholar] [CrossRef]
- Nattrass, G.S.; Banks, R.G.; Pitchford, W.S. The effect of telomere length variation on lifetime productivity traits in sheep. In Proceedings of the Association for the Advancement of Animal Breeding and Genetics Conference, Perth, WA, Australia, 19–21 July 2011; pp. 247–250. [Google Scholar]
- Froy, H.; Bird, E.J.; Wilbourn, R.V.; Fairlie, J.; Underwood, S.L.; Salvo-Chirnside, E.; Pilkington, J.G.; Berenos, C.; Pemberton, J.M.; Nussey, D.H. No evidence for parental age effects on offspring leukocyte telomere length in free-living Soay sheep. Sci. Rep. 2017, 7, 9991. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef]
- Cohen, J. A power primer. Psychol. Bull. 1992, 112, 155–159. [Google Scholar] [CrossRef]
- Daios, S.; Anogeianaki, A.; Kaiafa, G.; Kontana, A.; Veneti, S.; Gogou, C.; Karlafti, E.; Pilalas, D.; Kanellos, I.; Savopoulos, C. Telomere Length as a Marker of Biological Aging: A Critical Review of Recent Literature. Curr. Med. Chem. 2022, 29, 5478–5495. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, H. Mitochondrial DNA Copy Number and Developmental Origins of Health and Disease (DOHaD). Int. J. Mol. Sci. 2021, 22, 6634. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Lewis, C.J.; Dixit, B.; O’Connor, M.S.; Stolzing, A.; Boominathan, A. The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics. Biomedicines 2022, 10, 490. [Google Scholar] [CrossRef]
- Kolbe, I.; Brehm, N.; Oster, H. Interplay of central and peripheral circadian clocks in energy metabolism regulation. J. Neuroendocrinol. 2019, 31, e12659. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Johnston, J.D.; Messager, S.; Hazlerigg, D.; Lincoln, G. Photoperiod regulates clock gene rhythms in the ovine liver. Gen. Comp. Endocrinol. 2005, 142, 357–363. [Google Scholar] [CrossRef]
- Varcoe, T.J.; Gatford, K.L.; Voultsios, A.; Salkeld, M.D.; Boden, M.J.; Rattanatray, L.; Kennaway, D.J. Rapidly alternating photoperiods disrupt central and peripheral rhythmicity and decrease plasma glucose, but do not affect glucose tolerance or insulin secretion in sheep. Exp. Physiol. 2014, 99, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; la Fleur, S.; Fliers, E. Circadian control of glucose metabolism. Mol. Metab. 2014, 3, 372–383. [Google Scholar] [CrossRef]
- Gachon, F.; Loizides-Mangold, U.; Petrenko, V.; Dibner, C. Glucose Homeostasis: Regulation by Peripheral Circadian Clocks in Rodents and Humans. Endocrinology 2017, 158, 1074–1084. [Google Scholar] [CrossRef]
- Bu, Y.R.; Chen, S.; Ruan, M.C.; Wu, L.B.; Wang, H.L.; Li, N.; Zhao, X.J.; Yu, X.L.; Liu, Z.G. Per1/Per2 double knockout transcriptome analysis reveals circadian regulation of hepatic lipid metabolism. Food Sci. Hum. Wellness 2023, 12, 1716–1729. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhou, M.; Wang, S.; Hua, Z.; Zhang, J. Loss of mPer2 increases plasma insulin levels by enhanced glucose-stimulated insulin secretion and impaired insulin clearance in mice. FEBS Lett. 2012, 586, 1306–1311. [Google Scholar] [CrossRef]
- Shu, Y.Y.; Gao, W.K.; Chu, H.K.; Yang, L.; Pan, X.L.; Ye, J. Attenuation by Time-Restricted Feeding of High-Fat and High-Fructose Diet-Induced NASH in Mice Is Related to Per2 and Ferroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 8063897. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Tseng, H.L.; Shieh, K.R. Circadian-clock system in mouse liver affected by insulin resistance. Chronobiol. Int. 2013, 30, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Wasselin, T.; Zahn, S.; Maho, Y.L.; Dorsselaer, A.V.; Raclot, T.; Bertile, F. Exacerbated oxidative stress in the fasting liver according to fuel partitioning. Proteomics 2014, 14, 1905–1921. [Google Scholar] [CrossRef] [PubMed]
- Tal, Y.; Chapnik, N.; Froy, O. Non-obesogenic doses of fatty acids modulate the functionality of the circadian clock in the liver. Cell. Mol. Life Sci. 2019, 76, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.; Loudon, A.S.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Kennaway, D.J.; Owens, J.A.; Voultsios, A.; Boden, M.J.; Varcoe, T.J. Metabolic homeostasis in mice with disrupted Clock gene expression in peripheral tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1528–R1537. [Google Scholar] [CrossRef]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef]
- Sookoian, S.; Castano, G.; Gemma, C.; Gianotti, T.F.; Pirola, C.J. Common genetic variations in CLOCK transcription factor are associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 4242–4248. [Google Scholar] [CrossRef]
- Yuan, G.; Hua, B.; Cai, T.; Xu, L.; Li, E.; Huang, Y.; Sun, N.; Yan, Z.; Lu, C.; Qian, R. Clock mediates liver senescence by controlling ER stress. Aging 2017, 9, 2647–2665. [Google Scholar] [CrossRef]
- Li, T.; Cao, R.; Xia, R.; Xia, Z. Effects of 72 hours sleep deprivation on liver circadian clock gene expression and oxidative stress in rats. Yangtze Med. 2017, 1, 194–201. [Google Scholar] [CrossRef]
- Bertolucci, C.; Cavallari, N.; Colognesi, I.; Aguzzi, J.; Chen, Z.; Caruso, P.; Foa, A.; Tosini, G.; Bernardi, F.; Pinotti, M. Evidence for an overlapping role of CLOCK and NPAS2 transcription factors in liver circadian oscillators. Mol. Cell. Biol. 2008, 28, 3070–3075. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.S. Reactive oxygen species-induced changes in glucose and lipid metabolism contribute to the accumulation of cholesterol in the liver during aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 2021, 3, 100301. [Google Scholar] [CrossRef] [PubMed]
- Laish, I.; Mannasse-Green, B.; Hadary, R.; Biron-Shental, T.; Konikoff, F.M.; Amiel, A.; Kitay-Cohen, Y. Telomere Dysfunction in Nonalcoholic Fatty Liver Disease and Cryptogenic Cirrhosis. Cytogenet. Genome Res. 2016, 150, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Zanet, Y.I.; Hashem, E.M.; Dessouky, M.N.; Megalla, M.H.; Dessouky, I.S. Study of serum bisphenol-A and the mRNA of galactosidase beta 1 and tumor necrosis factor alpha in Egyptian patients with type 2 diabetes mellitus. Egypt. J. Intern. Med. 2023, 35. [Google Scholar] [CrossRef]
- Koh, Y.C.; Kuo, L.H.; Chang, Y.Y.; Tung, Y.C.; Lo, Y.C.; Pan, M.H. Modulatory Effect of Fermented Black Soybean and Adlay on Gut Microbiota Contributes to Healthy Aging. Mol. Nutr. Food Res. 2023, 67, e2200700. [Google Scholar] [CrossRef] [PubMed]
- Baboota, R.K.; Rawshani, A.; Bonnet, L.; Li, X.; Yang, H.; Mardinoglu, A.; Tchkonia, T.; Kirkland, J.L.; Hoffmann, A.; Dietrich, A.; et al. BMP4 and Gremlin 1 regulate hepatic cell senescence during clinical progression of NAFLD/NASH. Nat. Metab. 2022, 4, 1007–1021. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Teng, Y.C.; Lin, C.H.; Tsai, T.F. Cisd2 Protects the Liver from Oxidative Stress and Ameliorates Western Diet-Induced Nonalcoholic Fatty Liver Disease. Antioxidants 2021, 10, 559. [Google Scholar] [CrossRef]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Shen, Z.Q.; Lin, C.C.; Lu, C.K.; Tsai, T.F. Rejuvenation: Turning Back Time by Enhancing CISD2. Int. J. Mol. Sci. 2022, 23, 14014. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Lin, C.H.; Tsai, T.F. Cisd2 slows down liver aging and attenuates age-related metabolic dysfunction in male mice. Aging Cell 2021, 20, e13523. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Kuchel, G.A.; Melzer, D. ApoE e2 and aging-related outcomes in 379,000 UK Biobank participants. Aging 2020, 12, 12222–12233. [Google Scholar] [CrossRef] [PubMed]
- Bian, A.; Neyra, J.A.; Zhan, M.; Hu, M.C. Klotho, stem cells, and aging. Clin. Interv. Aging 2015, 10, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef]
- Lu, W.; Mei, J.; Yang, J.; Wu, Z.; Liu, J.; Miao, P.; Chen, Y.; Wen, Z.; Zhao, Z.; Kong, H.; et al. ApoE deficiency promotes non-alcoholic fatty liver disease in mice via impeding AMPK/mTOR mediated autophagy. Life Sci. 2020, 252, 117601. [Google Scholar] [CrossRef]
- Ang, L.S.; Cruz, R.P.; Hendel, A.; Granville, D.J. Apolipoprotein E, an important player in longevity and age-related diseases. Exp. Gerontol. 2008, 43, 615–622. [Google Scholar] [CrossRef]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Jiang, W.; You, N.; Huang, X.; Li, Y.; Peng, X.; Dong, R.; Wang, Z.; Zhu, Y.; Wu, K.; et al. Soluble Klotho Improves Hepatic Glucose and Lipid Homeostasis in Type 2 Diabetes. Mol. Ther. Methods Clin. Dev. 2020, 18, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Landry, T.; Shookster, D.; Huang, H. Circulating alpha-klotho regulates metabolism via distinct central and peripheral mechanisms. Metabolism 2021, 121, 154819. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Mao, Y.; Lu, M.; Liu, X.; Sun, Y.; Li, Z.; Li, Y.; Ding, Y.; Zhang, J.; Hong, J.; et al. Association between serum Klotho levels and the prevalence of diabetes among adults in the United States. Front. Endocrinol. 2022, 13, 1005553. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Kim, S.S.; Lee, J.S.; Kim, I.J.; Song, S.H.; Cha, S.K.; Park, K.S.; Kang, J.S.; Chung, C.H. Soluble alpha-klotho as a novel biomarker in the early stage of nephropathy in patients with type 2 diabetes. PLoS ONE 2014, 9, e102984. [Google Scholar] [CrossRef]
- Chi, Z.; Teng, Y.; Liu, Y.; Gao, L.; Yang, J.; Zhang, Z. Association between klotho and non-alcoholic fatty liver disease and liver fibrosis based on the NHANES 2007-2016. Ann. Hepatol. 2023, 28, 101125. [Google Scholar] [CrossRef]
- Jing, Y.; Sun, Q.; Xiong, X.; Meng, R.; Tang, S.; Cao, S.; Bi, Y.; Zhu, D. Hepatocyte growth factor alleviates hepatic insulin resistance and lipid accumulation in high-fat diet-fed mice. J. Diabetes Investig. 2019, 10, 251–260. [Google Scholar] [CrossRef]
- Kim, S.Y. Noninvasive markers for the diagnosis of nonalcoholic Fatty liver disease. Endocrinol. Metab. 2013, 28, 280–282. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Shahni, R.; Rodriguez-de-Ledesma, A.; Laftah, A.; Cunningham, P. Mitochondrial DNA as a non-invasive biomarker: Accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem. Biophys. Res. Commun. 2011, 412, 1–7. [Google Scholar] [CrossRef]
- Kamfar, S.; Alavian, S.M.; Houshmand, M.; Yadegarazari, R.; Seifi Zarei, B.; Khalaj, A.; Shabab, N.; Saidijam, M. Liver Mitochondrial DNA Copy Number and Deletion Levels May Contribute to Nonalcoholic Fatty Liver Disease Susceptibility. Hepat. Mon. 2016, 16, e40774. [Google Scholar] [CrossRef] [PubMed]
- Gavia-Garcia, G.; Rosado-Perez, J.; Arista-Ugalde, T.L.; Aguiniga-Sanchez, I.; Santiago-Osorio, E.; Mendoza-Nunez, V.M. Telomere Length and Oxidative Stress and Its Relation with Metabolic Syndrome Components in the Aging. Biology 2021, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, Y.; Li, H.; Yin, F.; Ge, M.; Hu, L.; Zi, M.; Qin, Z.; He, Y. Telomere length is maternally inherited and associated with lipid metabolism in Chinese population. Aging 2022, 14, 354–367. [Google Scholar] [CrossRef]
- Erdem, H.B.; Bahsi, T.; Ergun, M.A. Function of telomere in aging and age related diseases. Environ. Toxicol. Pharmacol. 2021, 85, 103641. [Google Scholar] [CrossRef]
- Shin, H.K.; Park, J.H.; Yu, J.H.; Jin, Y.J.; Suh, Y.J.; Lee, J.W.; Kim, W.; Korean Nonalcoholic Fatty Liver Study, G. Association between telomere length and hepatic fibrosis in non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, 18004. [Google Scholar] [CrossRef]
- Kim, D.; Li, A.A.; Ahmed, A. Leucocyte telomere shortening is associated with nonalcoholic fatty liver disease-related advanced fibrosis. Liver Int. 2018, 38, 1839–1848. [Google Scholar] [CrossRef]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef]
- Kalyesubula, M.; Mopuri, R.; Rosov, A.; Alon, T.; Edery, N.; Moallem, U.; Dvir, H. Hyperglycemia-stimulating diet induces liver steatosis in sheep. Sci. Rep. 2020, 10, 12189. [Google Scholar] [CrossRef] [PubMed]
- Vassilatou, E. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J. Gastroenterol. 2014, 20, 8351–8363. [Google Scholar] [CrossRef] [PubMed]
- Puttabyatappa, M.; Andriessen, V.; Mesquitta, M.; Zeng, L.; Pennathur, S.; Padmanabhan, V. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Mediators of Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology 2017, 158, 2783–2798. [Google Scholar] [CrossRef] [PubMed]
- Hogg, K.; Wood, C.; McNeilly, A.S.; Duncan, W.C. The in utero programming effect of increased maternal androgens and a direct fetal intervention on liver and metabolic function in adult sheep. PLoS ONE 2011, 6, e24877. [Google Scholar] [CrossRef] [PubMed]
- Gounden, V.; Warasally, M.Z.; Magwai, T.; Naidoo, R.; Chuturgoon, A. A pilot study: Relationship between Bisphenol A, Bisphenol A glucuronide and sex steroid hormone levels in cord blood in A South African population. Reprod. Toxicol. 2021, 100, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, K.; Park, J.; Moon, H.-B.; Choi, G.; Lee, J.J.; Suh, E.; Kim, H.-J.; Eun, S.-H.; Kim, G.-H.; et al. Bisphenol A distribution in serum, urine, placenta, breast milk, and umbilical cord serum in a birth panel of mother–neonate pairs. Sci. Total Environ. 2018, 626, 1494–1501. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Pennathur, S.; Kannan, K.; Patisaul, H.B.; Dolinoy, D.C.; Zeng, L.; Padmanabhan, V. Impact of Gestational Bisphenol A on Oxidative Stress and Free Fatty Acids: Human Association and Interspecies Animal Testing Studies. Endocrinology 2015, 156, 911–922. [Google Scholar] [CrossRef]
- Mammadov, E.; Uncu, M.; Dalkan, C. High Prenatal Exposure to Bisphenol A Reduces Anogenital Distance in Healthy Male Newborns. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 25–29. [Google Scholar] [CrossRef]
- Corrales, J.; Kristofco, L.A.; Steele, W.B.; Yates, B.S.; Breed, C.S.; Williams, E.S.; Brooks, B.W. Global Assessment of Bisphenol A in the Environment: Review and Analysis of Its Occurrence and Bioaccumulation. Dose-Response 2015, 13, 1559325815598308. [Google Scholar] [CrossRef]
- Vasiljevic, T.; Harner, T. Bisphenol A and its analogues in outdoor and indoor air: Properties, sources and global levels. Sci. Total Environ. 2021, 789, 148013. [Google Scholar] [CrossRef]
- Wang, R.G.; Tan, T.J.; Liang, H.J.; Huang, Y.; Dong, S.J.; Wang, P.L.; Su, X.O. Occurrence and distribution of bisphenol compounds in different categories of animal feeds used in China. Emerg. Contam. 2021, 7, 179–186. [Google Scholar] [CrossRef]
- Alharbi, H.F.; Algonaiman, R.; Alduwayghiri, R.; Aljutaily, T.; Algheshairy, R.M.; Almutairi, A.S.; Alharbi, R.M.; Alfurayh, L.A.; Alshahwan, A.A.; Alsadun, A.F.; et al. Exposure to Bisphenol A Substitutes, Bisphenol S and Bisphenol F, and Its Association with Developing Obesity and Diabetes Mellitus: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 15918. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ding, K.; Huang, W.; Xu, F.; Lei, M.; Yue, R. Potential effects of bisphenol A on diabetes mellitus and its chronic complications: A narrative review. Heliyon 2023, 9, e16340. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, T.; Welz, P.S.; Benitah, S.A.; Koronowski, K.B. Collecting mouse livers for transcriptome analysis of daily rhythms. STAR Protoc. 2021, 2, 100539. [Google Scholar] [CrossRef]
- Dardente, H.; Fustin, J.M.; Hazlerigg, D.G. Transcriptional feedback loops in the ovine circadian clock. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2009, 153, 391–398. [Google Scholar] [CrossRef]
- Tang, J.; Ma, S.; Gao, Y.; Zeng, F.; Feng, Y.; Guo, C.; Hu, L.; Yang, L.; Chen, Y.; Zhang, Q.; et al. ANGPTL8 promotes adipogenic differentiation of mesenchymal stem cells: Potential role in ectopic lipid deposition. Front. Endocrinol. 2022, 13, 927763. [Google Scholar] [CrossRef]
- Schmitt, B.; Povinelli, L.; Crodian, J.; Casey, T.; Plaut, K. Circadian rhythms of ewes suckling singletons versus twins during the second week of lactation. BIOS 2014, 85, 207–217. [Google Scholar] [CrossRef]
- Xiong, Z.; Yang, F.; Xu, T.; Yang, Y.; Wang, F.; Zhou, G.; Wang, Q.; Guo, X.; Xing, C.; Bai, H.; et al. Selenium alleviates cadmium-induced aging via mitochondrial quality control in the livers of sheep. J. Inorg. Biochem. 2022, 232, 111818. [Google Scholar] [CrossRef]
- Zhang, G.M.; Zhang, T.T.; Jin, Y.H.; Liu, J.L.; Guo, Y.X.; Fan, Y.X.; El-Samahy, M.A.; Meng, F.X.; Wang, F.; Lei, Z.H. Effect of caloric restriction and subsequent re-alimentation on oxidative stress in the liver of Hu sheep ram lambs. Anim. Feed. Sci. Technol. 2018, 237, 68–77. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motta, G.; Thangaraj, S.V.; Padmanabhan, V. Developmental Programming: Impact of Prenatal Exposure to Bisphenol A on Senescence and Circadian Mediators in the Liver of Sheep. Toxics 2024, 12, 15. https://doi.org/10.3390/toxics12010015
Motta G, Thangaraj SV, Padmanabhan V. Developmental Programming: Impact of Prenatal Exposure to Bisphenol A on Senescence and Circadian Mediators in the Liver of Sheep. Toxics. 2024; 12(1):15. https://doi.org/10.3390/toxics12010015
Chicago/Turabian StyleMotta, Giuliana, Soundara Viveka Thangaraj, and Vasantha Padmanabhan. 2024. "Developmental Programming: Impact of Prenatal Exposure to Bisphenol A on Senescence and Circadian Mediators in the Liver of Sheep" Toxics 12, no. 1: 15. https://doi.org/10.3390/toxics12010015
APA StyleMotta, G., Thangaraj, S. V., & Padmanabhan, V. (2024). Developmental Programming: Impact of Prenatal Exposure to Bisphenol A on Senescence and Circadian Mediators in the Liver of Sheep. Toxics, 12(1), 15. https://doi.org/10.3390/toxics12010015