
Plasma Concentrations of High Mobility Group Box 1 Proteins and Soluble Receptors for Advanced Glycation End-Products Are Relevant Biomarkers of Cognitive Impairment in Alcohol Use Disorder: A Pilot Study

 ,
,  , ,
, ,  , , ,
, , , 
 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Recruitment and Screening of Participants
2.3. Psychiatric and Neuropsychological Evaluation
2.4. Sample Collection
2.5. Protein Quantification and Immunoblotting for sRAGE
2.6. Oxidative Stress Analysis
2.7. Human High-Mobility Group Box 1 Protein ELISA
2.8. Apolipoprotein D ELISA
2.9. Human Nuclear Factor Erythroid 2-Related Factor 2 ELISA
2.10. Statistical Analysis
3. Results
3.1. Sociodemographic and Clinical Characteristics of AUD, AD, and Control Groups
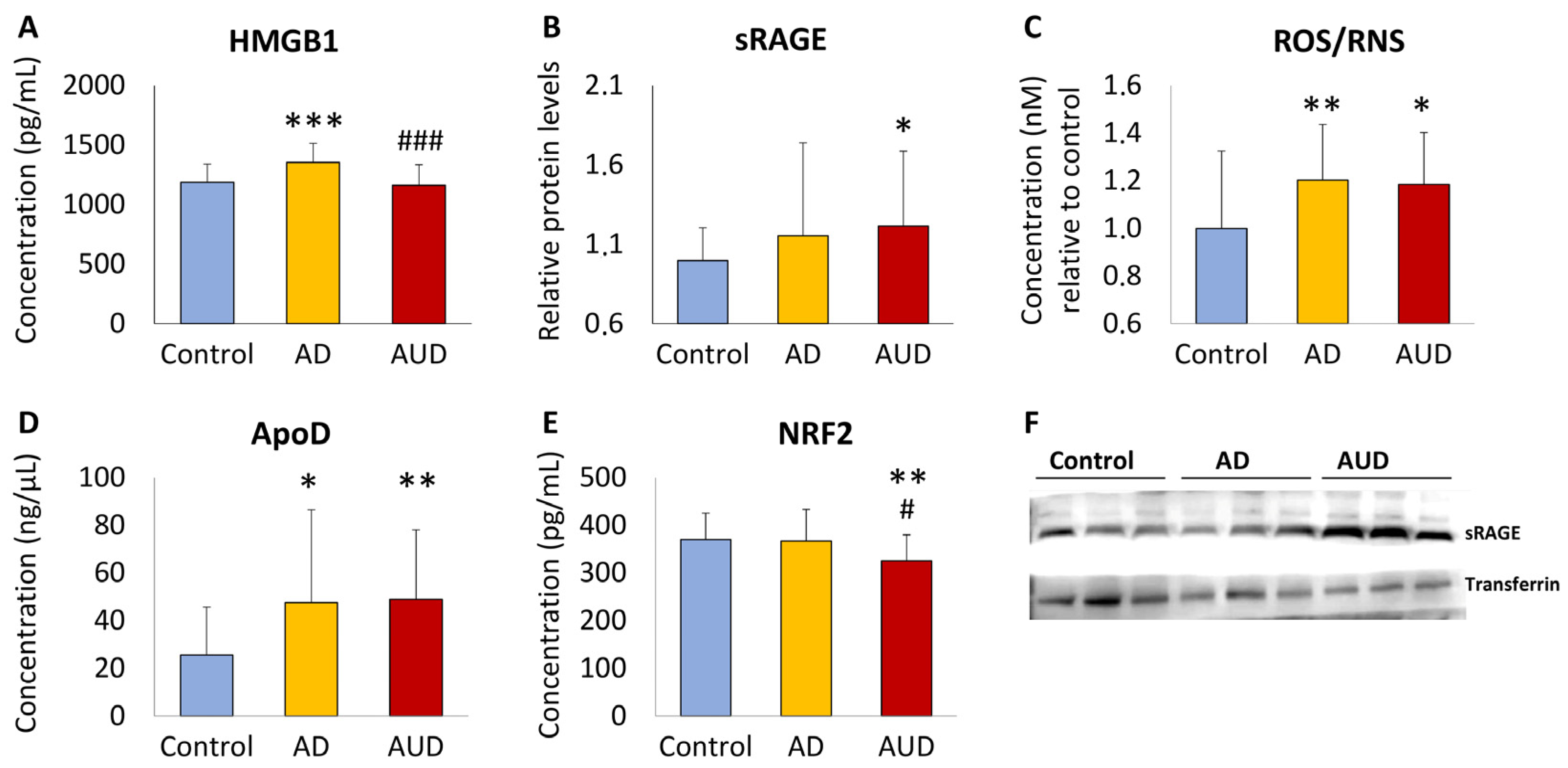
3.2. Cognitive Impairment in AUD Patients Alters Plasma Concentrations of HMGB1, RAGE, ROS/RNS, ApoD, and NRF2 Compared to AD and Control Groups
3.3. Comorbid Psychiatric Disorders Alter the Effect of Cognitive Impairment on Plasma Concentrations of sRAGE in AUD Patients
3.4. Correlation Analysis of HMGB1, sRAGE, ROS/RNS, ApoD, and NRF2 According to Variables of AUD Severity
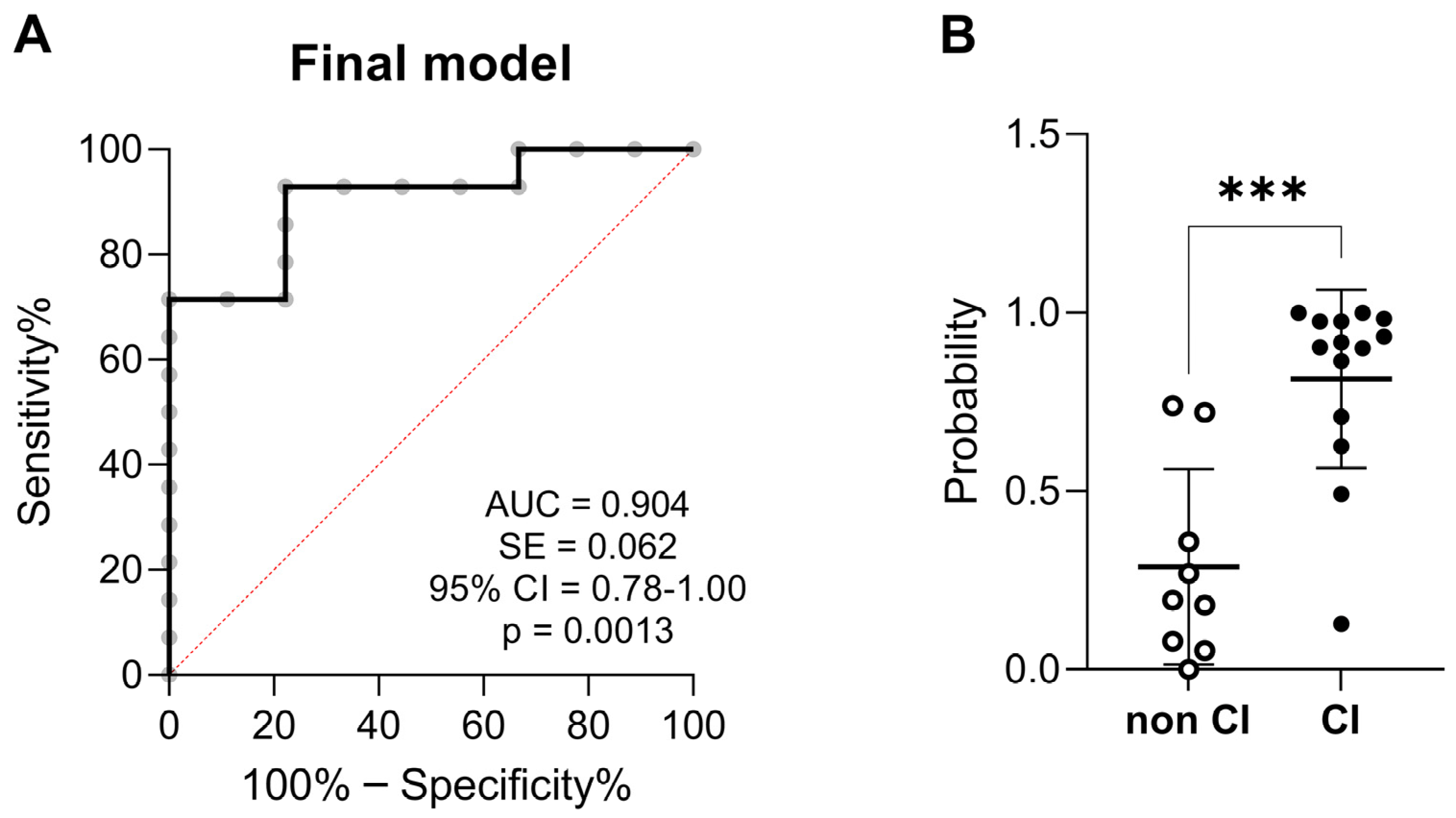
3.5. Predictive Variables of Cognitive Impairment in AUD Patients
4. Discussion
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NIAAA. Alcohol Facts and Statistics. The National Institute on Alcohol Abuse and Alcoholism. Available online: https://www.niaaa.nih.gov/sites/default/files/AlcoholFactsAndStats.pdf (accessed on 25 September 2023).
- Brust, J.C. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar] [CrossRef]
- Xu, G.; Liu, X.; Yin, Q.; Zhu, W.; Zhang, R.; Fan, X. Alcohol consumption and transition of mild cognitive impairment to dementia. Psychiatry Clin. Neurosci. 2009, 63, 43–49. [Google Scholar] [CrossRef]
- Hayes, V.; Demirkol, A.; Ridley, N.; Withall, A.; Draper, B. Alcohol-related cognitive impairment: Current trends and future perspectives. Neurodegener. Dis. Manag. 2016, 6, 509–523. [Google Scholar] [CrossRef]
- Visontay, R.; Rao, R.T.; Mewton, L. Alcohol use and dementia: New research directions. Curr. Opin. Psychiatry 2021, 34, 165–170. [Google Scholar] [CrossRef]
- Sachdeva, A.; Chandra, M.; Choudhary, M.; Dayal, P.; Anand, K.S. Alcohol-Related Dementia and Neurocognitive Impairment: A Review Study. Int. J. High Risk Behav. Addict. 2016, 5, e27976. [Google Scholar] [CrossRef]
- Wiegmann, C.; Mick, I.; Brandl, E.J.; Heinz, A.; Gutwinski, S. Alcohol and Dementia—What is the Link? A Systematic Review. Neuropsychiatr. Dis. Treat. 2020, 16, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.V.; Steinberg, R.A.; Han, D.; Sumbria, R.K. Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease-Evidence from Experimental Studies. Int. J. Mol. Sci. 2023, 24, 9492. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Lippai, D. Converging actions of alcohol on liver and brain immune signaling. Int. Rev. Neurobiol. 2014, 118, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Yang, Q.; Joshi, R.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhou, X.; Tan, X.; Huang, X.; Yuan, L.; Zhang, Z.; Yang, Y.; Xu, M.; Wan, Y.; Li, Z. The Status of Oxidative Stress in Patients with Alcohol Dependence: A Meta-Analysis. Antioxidants 2022, 11, 1919. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Advanced glycation endproducts: From precursors to RAGE: Round and round we go. Amino Acids 2012, 42, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Corica, D.; Pepe, G.; Currò, M.; Aversa, T.; Tropeano, A.; Ientile, R.; Wasniewska, M. Methods to investigate advanced glycation end-product and their application in clinical practice. Methods 2022, 203, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Coleman, L.G., Jr.; Macht, V.A.; Vetreno, R.P. Targeting Persistent Changes in Neuroimmune and Epigenetic Signaling in Adolescent Drinking to Treat Alcohol Use Disorder in Adulthood. Pharmacol. Rev. 2023, 75, 380–396. [Google Scholar] [CrossRef]
- Geroldi, D.; Falcone, C.; Emanuele, E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr. Med. Chem. 2006, 13, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Münch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xie, J.W.; Xu, Y.; Wang, T.; Cai, J.H.; Wang, X.; Zhao, B.L.; An, L.; Wang, Z.Y. Trientine reduces BACE1 activity and mitigates amyloidosis via the AGE/RAGE/NF-κB pathway in a transgenic mouse model of Alzheimer’s disease. Antioxid. Redox Signal. 2013, 19, 2024–2039. [Google Scholar] [CrossRef]
- Byun, K.; Bayarsaikhan, D.; Bayarsaikhan, E.; Son, M.; Oh, S.; Lee, J.; Son, H.I.; Won, M.H.; Kim, S.U.; Song, B.J.; et al. Microglial AGE-albumin is critical in promoting alcohol-induced neurodegeneration in rats and humans. PLoS ONE 2014, 9, e104699. [Google Scholar] [CrossRef]
- Prasad, K.; Mishra, M. AGE-RAGE Stress, Stressors, and Antistressors in Health and Disease. Int. J. Angiol. 2018, 27, 1–12. [Google Scholar] [CrossRef]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef]
- Crews, F.T.; Vetreno, R.P. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 2016, 233, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, V.; Grilli, M. Every Cloud Has a Silver Lining: Proneurogenic Effects of Aβ Oligomers and HMGB-1 via Activation of the RAGE-NF-κB Axis. CNS Neurol. Disord. Drug Targets 2017, 16, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. Is there any evidence that AGE/sRAGE is a universal biomarker/risk marker for diseases? Mol. Cell. Biochem. 2019, 451, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, E.; D’Angelo, A.; Tomaino, C.; Binetti, G.; Ghidoni, R.; Politi, P.; Bernardi, L.; Maletta, R.; Bruni, A.C.; Geroldi, D. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch. Neurol. 2005, 62, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Ghidoni, R.; Benussi, L.; Glionna, M.; Franzoni, M.; Geroldi, D.; Emanuele, E.; Binetti, G. Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment. J. Neural. Transm. 2008, 115, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, R.; Lu, S.; Xia, W.; Cai, R.; Sun, H.; Wang, S. RAGE and AGEs in Mild Cognitive Impairment of Diabetic Patients: A Cross-Sectional Study. PLoS ONE 2016, 11, e0145521. [Google Scholar] [CrossRef]
- Semchyshyn, H. Is carbonyl/AGE/RAGE stress a hallmark of the brain aging? Pflugers Arch. 2021, 473, 723–734. [Google Scholar] [CrossRef]
- Gasparotto, J.; Somensi, N.; Girardi, C.S.; Bittencourt, R.R.; de Oliveira, L.M.; Hoefel, L.P.; Scheibel, I.M.; Peixoto, D.O.; Moreira, J.C.F.; Outeiro, T.F.; et al. Is it all the RAGE? Defining the role of the receptor for advanced glycation end products in Parkinson’s disease. J. Neurochem. 2023. [Google Scholar] [CrossRef]
- Mo, J.; Hu, J.; Cheng, X. The role of high mobility group box 1 in neuroinflammatory related diseases. Biomed. Pharmacother. 2023, 161, 114541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Zeh, H.J.; Lotze, M.T. The multifunctional protein HMGB1: 50 years of discovery. Nat. Rev. Immunol. 2023, 23, 824–841. [Google Scholar] [CrossRef] [PubMed]
- Pilzweger, C.; Holdenrieder, S. Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagnostics 2015, 5, 219–253. [Google Scholar] [CrossRef] [PubMed]
- Staitieh, B.S.; Egea, E.E.; Fan, X.; Amah, A.; Guidot, D.M. Chronic Alcohol Ingestion Impairs Rat Alveolar Macrophage Phagocytosis via Disruption of RAGE Signaling. Am. J. Med. Sci. 2018, 355, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J.; QalyDays StudyGroup. Contribution of alcohol use disorders to the burden of dementia in France 2008-13: A nationwide retrospective cohort study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar] [CrossRef] [PubMed]
- Torrens, M.; Serrano, D.; Astals, M.; Pérez-Domínguez, G.; Martín-Santos, R. Diagnosing comorbid psychiatric disorders in substance abusers: Validity of the Spanish versions of the Psychiatric Research Interview for Substance and Mental Disorders and the Structured Clinical Interview for DSM-IV. Am. J. Psychiatry 2004, 161, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Hasin, D.S.; O’Brien, C.P.; Auriacombe, M.; Borges, G.; Bucholz, K.; Budney, A.; Compton, W.M.; Crowley, T.; Ling, W.; Petry, N.M.; et al. DSM-5 criteria for substance use disorders: Recommendations and rationale. Am. J. Psychiatry 2013, 170, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Requena-Ocaña, N.; Araos, P.; Serrano-Castro, P.J.; Flores-López, M.; García-Marchena, N.; Oliver-Martos, B.; Ruiz, J.J.; Gavito, A.; Pavón, F.J.; Serrano, A.; et al. Plasma Concentrations of Neurofilament Light Chain Protein and Brain-Derived Neurotrophic Factor as Consistent Biomarkers of Cognitive Impairment in Alcohol Use Disorder. Int. J. Mol. Sci. 2023, 24, 1183. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dementia: A Public Health Priority (World Health Organization, 2012). Available online: https://www.who.int/publications/i/item/dementia-a-public-health-priority (accessed on 25 September 2023).
- Withall, A.; Draper, B.; Seeher, K.; Brodaty, H. The prevalence and causes of younger onset dementia in Eastern Sydney, Australia. Int. Psychogeriatr. 2014, 26, 1955–1965. [Google Scholar] [CrossRef]
- Woods, A.J.; Porges, E.C.; Bryant, V.E.; Seider, T.; Gongvatana, A.; Kahler, C.W.; de la Monte, S.; Monti, P.M.; Cohen, R.A. Current Heavy Alcohol Consumption is Associated with Greater Cognitive Impairment in Older Adults. Alcohol. Clin. Exp. Res. 2016, 40, 2435–2444. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflamm. 2012, 9, 5. [Google Scholar] [CrossRef]
- Ganfornina, M.D.; Do Carmo, S.; Martínez, E.; Tolivia, J.; Navarro, A.; Rassart, E.; Sanchez, D. ApoD, a glia-derived apolipoprotein, is required for peripheral nerve functional integrity and a timely response to injury. Glia 2010, 58, 1320–1334. [Google Scholar] [CrossRef]
- Fyfe-Desmarais, G.; Desmarais, F.; Rassart, É.; Mounier, C. Apolipoprotein D in Oxidative Stress and Inflammation. Antioxidants 2023, 12, 1027. [Google Scholar] [CrossRef]
- Vetreno, R.P.; Qin, L.; Crews, F.T. Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol. Dis. 2013, 59, 52–62. [Google Scholar] [CrossRef]
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol Use Disorder, Neurodegeneration, Alzheimer’s and Parkinson’s Disease: Interplay Between Oxidative Stress, Neuroimmune Response and Excitotoxicity. Front. Cell Neurosci. 2020, 14, 282. [Google Scholar] [CrossRef]
- Crews, F.T.; Fisher, R.P.; Qin, L.; Vetreno, R.P. HMGB1 neuroimmune signaling and REST-G9a gene repression contribute to ethanol-induced reversible suppression of the cholinergic neuron phenotype. Mol. Psychiatry 2023. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Stern, D.M.; Arnold, B.; Nawroth, P.P. Advanced glycation end product receptor-mediated cellular dysfunction. Ann. N. Y. Acad. Sci. 2005, 1043, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Fukami, A.; Adachi, H.; Yamagishi, S.; Matsui, T.; Ueda, S.; Nakamura, K.; Enomoto, M.; Otsuka, M.; Kumagae, S.; Nanjo, Y.; et al. Factors associated with serum high mobility group box 1 (HMGB1) levels in a general population. Metabolism 2009, 58, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Aryal, P.; Soni, P. RAGE Inhibitors in Neurodegenerative Diseases. Biomedicines 2023, 11, 1131. [Google Scholar] [CrossRef] [PubMed]
- Selvin, E.; Halushka, M.K.; Rawlings, A.M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Astor, B.C. sRAGE and risk of diabetes, cardiovascular disease, and death. Diabetes 2013, 62, 2116–2121. [Google Scholar] [CrossRef]
- Zgutka, K.; Tkacz, M.; Tomasiak, P.; Tarnowski, M. A Role for Advanced Glycation End Products in Molecular Ageing. Int. J. Mol. Sci. 2023, 24, 9881. [Google Scholar] [CrossRef]
- Requena-Ocaña, N.; Flores-Lopez, M.; Papaseit, E.; García-Marchena, N.; Ruiz, J.J.; Ortega-Pinazo, J.; Serrano, A.; Pavón-Morón, F.J.; Farré, M.; Suarez, J.; et al. Vascular Endothelial Growth Factor as a Potential Biomarker of Neuroinflammation and Frontal Cognitive Impairment in Patients with Alcohol Use Disorder. Biomedicines 2022, 10, 947. [Google Scholar] [CrossRef]
- García-Marchena, N.; Silva-Peña, D.; Martín-Velasco, A.I.; Villanúa, M.Á.; Araos, P.; Pedraz, M.; Maza-Quiroga, R.; Romero-Sanchiz, P.; Rubio, G.; Castilla-Ortega, E.; et al. Decreased plasma concentrations of BDNF and IGF-1 in abstinent patients with alcohol use disorders. PLoS ONE 2017, 12, e0187634. [Google Scholar] [CrossRef]
- Silva-Peña, D.; García-Marchena, N.; Alén, F.; Araos, P.; Rivera, P.; Vargas, A.; García-Fernández, M.I.; Martín-Velasco, A.I.; Villanúa, M.Á.; Castilla-Ortega, E.; et al. Alcohol-induced cognitive deficits are associated with decreased circulating levels of the neurotrophin BDNF in humans and rats. Addict. Biol. 2019, 24, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- García-Marchena, N.; Pizarro, N.; Pavón, F.J.; Martínez-Huélamo, M.; Flores-López, M.; Requena-Ocaña, N.; Araos, P.; Silva-Peña, D.; Suárez, J.; Santín, L.J.; et al. Potential association of plasma lysophosphatidic acid (LPA) species with cognitive impairment in abstinent alcohol use disorders outpatients. Sci. Rep. 2020, 10, 17163. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Guerri, C. Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit. Rev. Clin. Lab. Sci. 2011, 48, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Alam, M.R.; Kamal, M.A.; Seo, K.J.; Singh, L.R. AGE-RAGE axis culminates into multiple pathogenic processes: A central road to neurodegeneration. Front. Mol. Neurosci. 2023, 16, 1155175. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Qin, L.; Sheedy, D.; Vetreno, R.P.; Zou, J. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatry 2013, 73, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017, 122, 56–73. [Google Scholar] [CrossRef] [PubMed]
- García-Marchena, N.; Maza-Quiroga, R.; Serrano, A.; Barrios, V.; Requena-Ocaña, N.; Suárez, J.; Chowen, J.A.; Argente, J.; Rubio, G.; Torrens, M.; et al. Abstinent patients with alcohol use disorders show an altered plasma cytokine profile: Identification of both interleukin 6 and interleukin 17A as potential biomarkers of consumption and comorbid liver and pancreatic diseases. J. Psychopharmacol. 2020, 34, 1250–1260. [Google Scholar] [CrossRef]
- Haffner, S.M.; Applebaum-Bowden, D.; Wahl, P.W.; Hoover, J.J.; Warnick, G.R.; Albers, J.J.; Hazzard, W.R. Epidemiological correlates of high density lipoprotein subfractions, apolipoproteins A-I, A-II, and D, and lecithin cholesterol acyltransferase. Effects of smoking, alcohol, and adiposity. Arteriosclerosis 1985, 5, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Flatscher-Bader, T.; Wilce, P.A. The effect of alcohol and nicotine abuse on gene expression in the brain. Nutr. Res. Rev. 2009, 22, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Ben Khedher, M.R.; Haddad, M.; Laurin, D.; Ramassamy, C. Effect of APOE ε4 allele on levels of apolipoproteins E, J, and D, and redox signature in circulating extracellular vesicles from cognitively impaired with no dementia participants converted to Alzheimer’s disease. Alzheimer’s Dement. 2021, 13, e12231. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Total Sample (N = 76) | |||||
|---|---|---|---|---|---|
| Variables | Control Group (N = 25) | AD Group (N = 26) | AUD Group (N = 25) | p-Value | |
| Age (mean ± SD) | Years | 52.48 ± 2.26 | 77.07 ± 4.65 | 42.07 ± 12.45 | <0.001 a |
| Body mass index (mean ± SD) | Kg/m2 | 27.89 ± 3.90 | 27.69 ± 2.53 | 25.59 ± 5.87 | 0.107 b |
| Sex [N (%)] | Women Men | 8 17 | 14 12 | 7 18 | 0.053 c |
| Variables | AUD Group | |
|---|---|---|
| Substance use [N (%)] | Alcohol | 13 (52) |
| Alcohol + cocaine | 4 (16) | |
| Alcohol + cannabis | 3 (12) | |
| Alcohol + cocaine + cannabis | 5 (20) | |
| AUD severity (mean ± SD) | Age (years) of use onset | 14.85 ± 4.21 |
| Age (years) of dependence onset | 21.96 ± 6.99 | |
| Duration (years) of use | 15.18 ± 13.16 | |
| Duration (days) of abstinence | 475.72 ± 1152 | |
| Periods of abstinence | 1.85 ± 2.10 | |
| Comorbid psychiatric disorders [N (%)] | Mood | 14 (56) |
| Anxiety | 12 (48) | |
| Personality | 7 (28) | |
| Psychotic | 2 (8) | |
| Cognitive impairment (MoCA a) [N (%)] | Yes | 17 (68) |
| No | 8 (32) | |
| Variables | AUD Group | |
|---|---|---|
| Dementia etiology * [N (%)] | Degenerative | 4 (15.38) |
| Amnesic | 6 (23.08) | |
| Degenerative + amnesic | 9 (34.61) | |
| Unknown | 4 (15.38) | |
| Neurodegenerative probability * [N (%)] | High | 10 (38.46) |
| Medium | 5 (19.23) | |
| Low | 8 (30.77) | |
| Blessed dementia test [N (%)] | Severe-moderate | 5 (19.23) |
| Mild | 21 (80.77) | |
| Episodic memory test [N (%)] | Affected | 2 (7.69) |
| Mild | 17 (65.38) | |
| No | 7 (26.92) | |
| Cognitive impairment (MoCA) * [N (%)] | Yes | 19 (73.08) |
| No | 7 (26.92) | |
| Cognitive Impairment | AD | AUD | p-Value |
|---|---|---|---|
| MoCA score (mean ± SD) | 23.04 ± 3.24 | 26.00 ± 3.82 | <0.001 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez de Fonseca, F.; Medina-Paz, F.; Sapozhnikov, M.; Hurtado-Guerrero, I.; Rubio, L.; Martín-de-las-Heras, S.; Requena-Ocaña, N.; Flores-López, M.; Fernández-Arjona, M.d.M.; Rivera, P.; et al. Plasma Concentrations of High Mobility Group Box 1 Proteins and Soluble Receptors for Advanced Glycation End-Products Are Relevant Biomarkers of Cognitive Impairment in Alcohol Use Disorder: A Pilot Study. Toxics 2024, 12, 190. https://doi.org/10.3390/toxics12030190
Rodríguez de Fonseca F, Medina-Paz F, Sapozhnikov M, Hurtado-Guerrero I, Rubio L, Martín-de-las-Heras S, Requena-Ocaña N, Flores-López M, Fernández-Arjona MdM, Rivera P, et al. Plasma Concentrations of High Mobility Group Box 1 Proteins and Soluble Receptors for Advanced Glycation End-Products Are Relevant Biomarkers of Cognitive Impairment in Alcohol Use Disorder: A Pilot Study. Toxics. 2024; 12(3):190. https://doi.org/10.3390/toxics12030190
Chicago/Turabian StyleRodríguez de Fonseca, Fernando, Francisco Medina-Paz, Mira Sapozhnikov, Isaac Hurtado-Guerrero, Leticia Rubio, Stella Martín-de-las-Heras, Nerea Requena-Ocaña, María Flores-López, María del Mar Fernández-Arjona, Patricia Rivera, and et al. 2024. "Plasma Concentrations of High Mobility Group Box 1 Proteins and Soluble Receptors for Advanced Glycation End-Products Are Relevant Biomarkers of Cognitive Impairment in Alcohol Use Disorder: A Pilot Study" Toxics 12, no. 3: 190. https://doi.org/10.3390/toxics12030190
APA StyleRodríguez de Fonseca, F., Medina-Paz, F., Sapozhnikov, M., Hurtado-Guerrero, I., Rubio, L., Martín-de-las-Heras, S., Requena-Ocaña, N., Flores-López, M., Fernández-Arjona, M. d. M., Rivera, P., Serrano, A., Serrano, P., C. Zapico, S., & Suárez, J. (2024). Plasma Concentrations of High Mobility Group Box 1 Proteins and Soluble Receptors for Advanced Glycation End-Products Are Relevant Biomarkers of Cognitive Impairment in Alcohol Use Disorder: A Pilot Study. Toxics, 12(3), 190. https://doi.org/10.3390/toxics12030190