Involvement of ERK and Oxidative Stress in Airway Exposure to Cadmium Chloride Aggravates Airway Inflammation in Ovalbumin-Induced Asthmatic Mice


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals Model
2.2. Chemical Inhibitor Administration
2.3. Bronchoalveolar Lavage Fluid (BALF) and Histopathological Collection
2.4. MDA and GSH-Px Analysis
2.5. Immunoblotting
2.6. Statistical Analysis
3. Results
3.1. Effects of CdCl2 Exposure on the Allergic Asthma Model
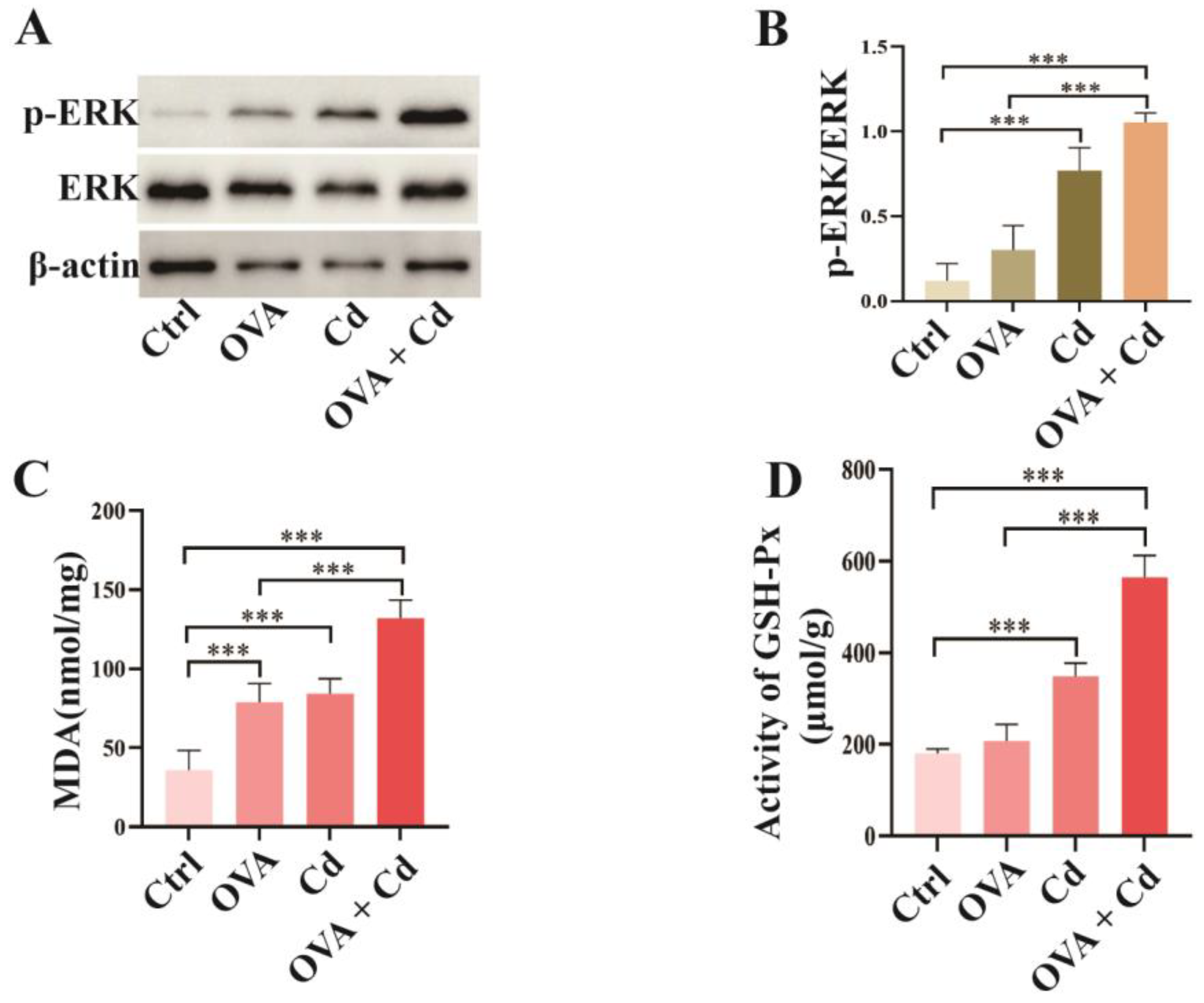
3.2. Airway Exposure to CdCl2 Enhances ERK Phosphorylation and Oxidative Stress in OVA-Induced Mice
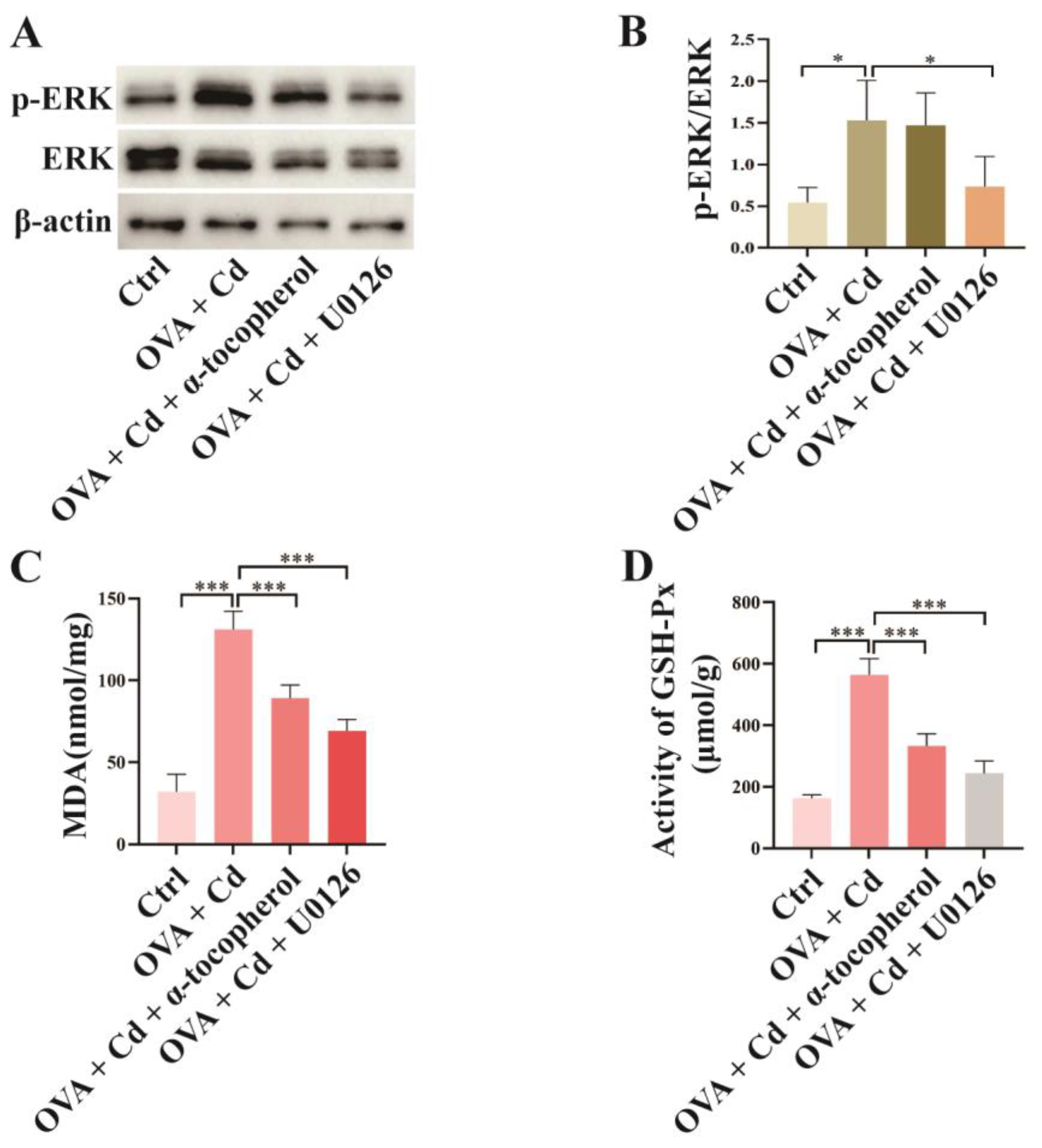
3.3. Effect of U0126 and α-Tocopherol Treatment on the OVA + CdCl2 Mouse Model
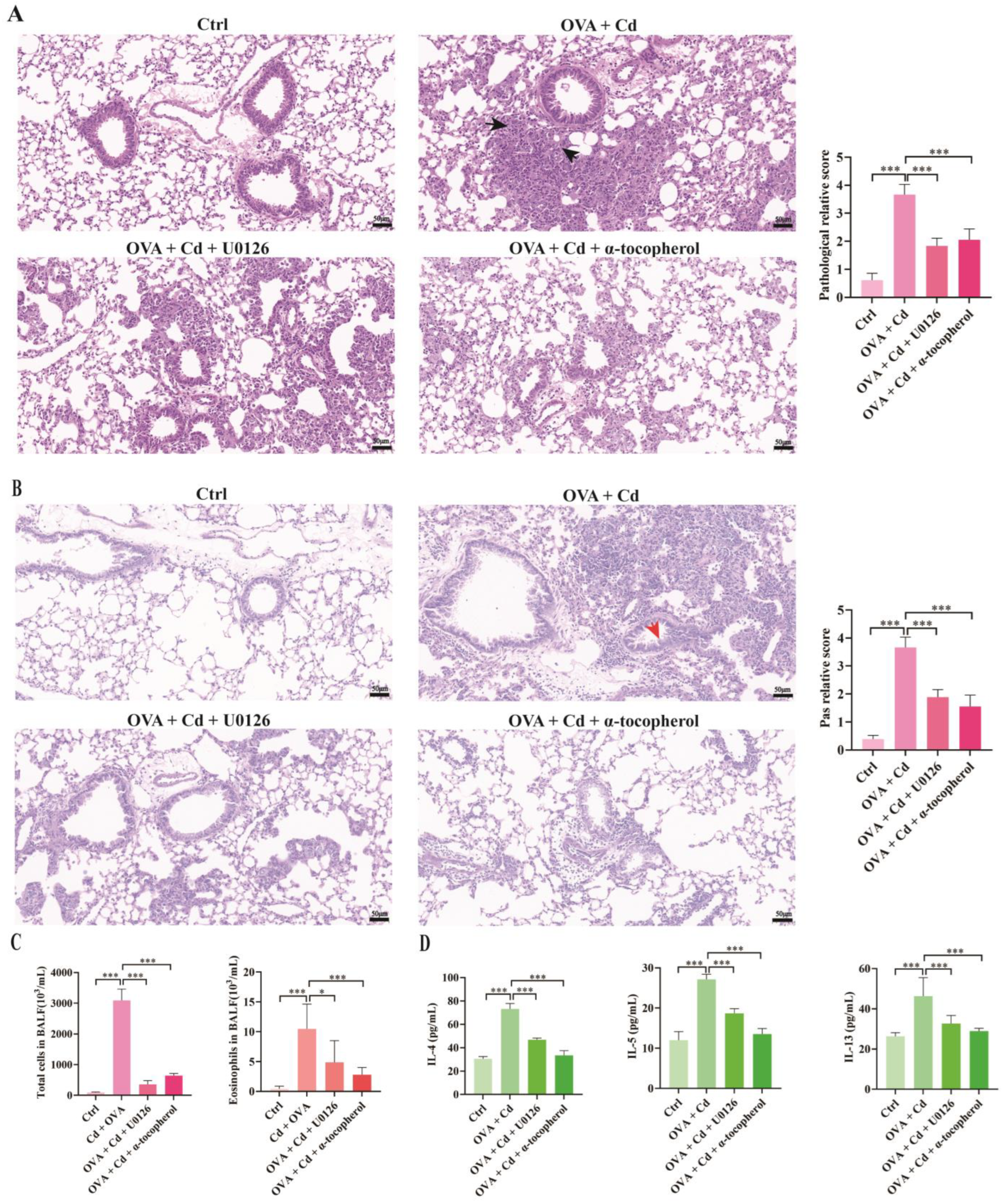
3.4. Role of ERK and Oxidative Stress in CdCl2-Exacerbated Asthma in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, R.L.; Grayson, M.H.; Strothman, K. Advances in asthma: New understandings of asthma’s natural history, risk factors, underlying mechanisms, and clinical management. J. Allergy Clin. Immunol. 2021, 148, 1430–1441. [Google Scholar] [CrossRef]
- Stikker, B.S.; Hendriks, R.W.; Stadhouders, R. Decoding the genetic and epigenetic basis of asthma. Allergy 2023, 78, 940–956. [Google Scholar] [CrossRef]
- Cai, L.M.; Wang, Q.S.; Luo, J.; Chen, L.G.; Zhu, R.L.; Wang, S.; Tang, C.H. Heavy metal contamination and health risk assessment for children near a large Cu-smelter in central China. Sci. Total Environ. 2019, 650, 725–733. [Google Scholar] [CrossRef]
- Peana, M.; Pelucelli, A.; Chasapis, C.T.; Perlepes, S.P.; Bekiari, V.; Medici, S.; Zoroddu, M.A. Biological Effects of Human Exposure to Environmental Cadmium. Biomolecules 2022, 13, 36. [Google Scholar] [CrossRef]
- Wang, W.J.; Peng, K.; Lu, X.; Zhu, Y.Y.; Li, Z.; Qian, Q.H.; Yao, Y.X.; Fu, L.; Wang, Y.; Huang, Y.C.; et al. Long-term cadmium exposure induces chronic obstructive pulmonary disease-like lung lesions in a mouse model. Sci. Total Environ. 2023, 879, 163073. [Google Scholar] [CrossRef]
- Knoell, D.L.; Wyatt, T.A. The adverse impact of cadmium on immune function and lung host defense. Semin. Cell Dev. Biol. 2021, 115, 70–76. [Google Scholar] [CrossRef]
- Kar, I.; Patra, A.K. Tissue Bioaccumulation and Toxicopathological Effects of Cadmium and Its Dietary Amelioration in Poultry-a Review. Biol. Trace Elem. Res. 2021, 199, 3846–3868. [Google Scholar] [CrossRef]
- Yi, L.; Shang, X.J.; Lv, L.; Wang, Y.; Zhang, J.; Quan, C.; Shi, Y.; Liu, Y.; Zhang, L. Cadmium-induced apoptosis of Leydig cells is mediated by excessive mitochondrial fission and inhibition of mitophagy. Cell Death Dis. 2022, 13, 928. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Zhang, M.; OuYang, H.; Li, M.; Jia, D.; Wang, R.; Zhou, W.; Liu, H.; Hu, Y.; et al. Cadmium exposure during puberty damages testicular development and spermatogenesis via ferroptosis caused by intracellular iron overload and oxidative stress in mice. Environ. Pollut. 2023, 325, 121434. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xun, P.; Nishijo, M.; He, K. Cadmium exposure and risk of lung cancer: A meta-analysis of cohort and case-control studies among general and occupational populations. J. Expo. Sci. Environ. Epidemiol. 2016, 26, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Filippini, T.; Torres, D.; Lopes, C.; Carvalho, C.; Moreira, P.; Naska, A.; Kasdagli, M.I.; Malavolti, M.; Orsini, N.; Vinceti, M. Cadmium exposure and risk of breast cancer: A dose-response meta-analysis of cohort studies. Environ. Int. 2020, 142, 105879. [Google Scholar] [CrossRef]
- Florez-Garcia, V.A.; Guevara-Romero, E.C.; Hawkins, M.M.; Bautista, L.E.; Jenson, T.E.; Yu, J.; Kalkbrenner, A.E. Cadmium exposure and risk of breast cancer: A meta-analysis. Environ. Res. 2023, 219, 115109. [Google Scholar] [CrossRef]
- Huang, X.; Xie, J.; Cui, X.; Zhou, Y.; Wu, X.; Lu, W.; Shen, Y.; Yuan, J.; Chen, W. Association between Concentrations of Metals in Urine and Adult Asthma: A Case-Control Study in Wuhan, China. PLoS ONE 2016, 11, e0155818. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Sun, T.; Han, Y.Y.; Rosser, F.; Forno, E.; Chen, W.; Celedón, J.C. Serum Cadmium and Lead, Current Wheeze, and Lung Function in a Nationwide Study of Adults in the United States. J. Allergy Clin. Immunol. Pract. 2019, 7, 2653–2660.e3. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.Y.; Kim, T.H.; Sheen, Y.H.; Lee, S.W.; An, J.; Kim, M.A.; Han, M.Y.; Yon, D.K. Serum heavy metal levels are associated with asthma, allergic rhinitis, atopic dermatitis, allergic multimorbidity, and airflow obstruction. J. Allergy Clin. Immunol. Pract. 2019, 7, 2912–2915.e2. [Google Scholar] [CrossRef]
- Wang, W.J.; Lu, X.; Li, Z.; Peng, K.; Zhan, P.; Fu, L.; Wang, Y.; Zhao, H.; Wang, H.; Xu, D.X.; et al. Early-life cadmium exposure elevates susceptibility to allergic asthma in ovalbumin-sensitized and challenged mice. Ecotoxicol. Environ. Saf. 2023, 255, 114799. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Liu, S.; Pyles, J.M.; Lapi, S.E.; Saleem, K.; Antony, V.B.; Gonzalez, M.L.; Crossman, D.K.; Carter, A.B. Impaired PPARγ activation by cadmium exacerbates infection-induced lung injury. JCI Insight 2023, 8, e166608. [Google Scholar] [CrossRef]
- Cao, X.; Fu, M.; Bi, R.; Zheng, X.; Fu, B.; Tian, S.; Liu, C.; Li, Q.; Liu, J. Cadmium induced BEAS-2B cells apoptosis and mitochondria damage via MAPK signaling pathway. Chemosphere 2021, 263, 128346. [Google Scholar] [CrossRef]
- Alruhaimi, R.S.; Hassanein, E.H.M.; Bin-Jumah, M.N.; Mahmoud, A.M. Cadmium-induced lung injury is associated with oxidative stress, apoptosis, and altered SIRT1 and Nrf2/HO-1 signaling; protective role of the melatonin agonist agomelatine. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 2335–2345. [Google Scholar] [CrossRef]
- Yu, F.; Sun, Y.; Yu, J.; Ding, Z.; Wang, J.; Zhang, L.; Zhang, T.; Bai, Y.; Wang, Y. ORMDL3 is associated with airway remodeling in asthma via the ERK/MMP-9 pathway. Mol. Med. Rep. 2017, 15, 2969–2976. [Google Scholar] [CrossRef]
- Wei, M.; Xie, X.; Chu, X.; Yang, X.; Guan, M.; Wang, D. Dihydroartemisinin suppresses ovalbumin-induced airway inflammation in a mouse allergic asthma model. Immunopharmacol. Immunotoxicol. 2013, 35, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Michaeloudes, C.; Abubakar-Waziri, H.; Lakhdar, R.; Raby, K.; Dixey, P.; Adcock, I.M.; Mumby, S.; Bhavsar, P.K.; Chung, K.F. Molecular mechanisms of oxidative stress in asthma. Mol. Aspects Med. 2022, 85, 101026. [Google Scholar] [CrossRef]
- Hu, X.; Deng, S.; Luo, L.; Jiang, Y.; Ge, H.; Yin, F.; Zhang, Y.; Zhang, D.; Li, X.; Feng, J. GLCCI1 Deficiency Induces Glucocorticoid Resistance via the Competitive Binding of IRF1:GRIP1 and IRF3:GRIP1 in Asthma. Front. Med. 2021, 8, 686493. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, S.H.; Jeon, D.; Kim, H.Y.; Han, J.Y.; Kim, B.; Lee, K. Low-dose cadmium exposure exacerbates polyhexamethylene guanidine-induced lung fibrosis in mice. J. Toxicol. Environ. Health A 2018, 81, 384–396. [Google Scholar] [CrossRef]
- Zou, M.; Zhang, G.; Zou, J.; Liu, Y.; Liu, B.; Hu, X.; Cheng, Z. Inhibition of the ERK1/2-ubiquitous calpains pathway attenuates experimental pulmonary fibrosis in vivo and in vitro. Exp. Cell Res. 2020, 391, 111886. [Google Scholar] [CrossRef]
- Bao, A.; Yang, H.; Ji, J.; Chen, Y.; Bao, W.; Li, F.; Zhang, M.; Zhou, X.; Li, Q.; Ben, S. Involvements of p38 MAPK and oxidative stress in the ozone-induced enhancement of AHR and pulmonary inflammation in an allergic asthma model. Respir. Res. 2017, 18, 216. [Google Scholar] [CrossRef]
- Pei, C.; Jia, N.; Wang, Y.; Zhao, S.; Shen, Z.; Shi, S.; Huang, D.; Wu, Y.; Wang, X.; Li, S.; et al. Notoginsenoside R1 protects against hypobaric hypoxia-induced high-altitude pulmonary edema by inhibiting apoptosis via ERK1/2-P90rsk-BAD ignaling pathway. Eur. J. Pharmacol. 2023, 959, 176065. [Google Scholar] [CrossRef]
- Mabalirajan, U.; Aich, J.; Leishangthem, G.D.; Sharma, S.K.; Dinda, A.K.; Ghosh, B. Effects of vitamin E on mitochondrial dysfunction and asthma features in an experimental allergic murine model. J. Appl. Physiol. 2009, 107, 1285–1292. [Google Scholar] [CrossRef]
- Xu, X.; Xu, J.F.; Zheng, G.; Lu, H.W.; Duan, J.L.; Rui, W.; Guan, J.H.; Cheng, L.Q.; Yang, D.D.; Wang, M.C.; et al. CARD9(S12N) facilitates the production of IL-5 by alveolar macrophages for the induction of type 2 immune responses. Nat. Immunol. 2018, 19, 547–560. [Google Scholar] [CrossRef]
- Cai, R.; Gong, X.; Li, X.; Jiang, Y.; Deng, S.; Tang, J.; Ge, H.; Wu, C.; Tang, H.; Wang, G.; et al. Dectin-1 aggravates neutrophil inflammation through caspase-11/4-mediated macrophage pyroptosis in asthma. Respir. Res. 2024, 25, 119. [Google Scholar] [CrossRef] [PubMed]
- Surolia, R.; Karki, S.; Kim, H.; Yu, Z.; Kulkarni, T.; Mirov, S.B.; Carter, A.B.; Rowe, S.M.; Matalon, S.; Thannickal, V.J.; et al. Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L280–L292. [Google Scholar] [CrossRef] [PubMed]
- Marzec, J.M.; Nadadur, S.S. Inflammation resolution in environmental pulmonary health and morbidity. Toxicol. Appl. Pharmacol. 2022, 449, 116070. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.C.; Polosa, R.; Sin, D.D. Cigarette Smoking and Asthma. J. Allergy Clin. Immunol. Pract. 2022, 10, 2783–2797. [Google Scholar] [CrossRef] [PubMed]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public. Health 2018, 15, 1033. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.E.; Church, T.L.; Chism, D.D.; Panettieri, R.A., Jr.; Shore, S.A. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: A role for ERK. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L847–L853. [Google Scholar] [CrossRef]
- Devi, K.; Soni, S.; Tripathi, V.; Pandey, R.; Moharana, B. Ethanolic Extract of Tridax procumbens Mitigates Pulmonary Inflammation via Inhibition of NF-κB/p65/ERK Mediated Signalling in an Allergic Asthma Model. Phytomedicine 2022, 99, 154008. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.M.; Choi, S.H.; Jee, W.; Lee, S.H.; Park, D.; Jung, J.H.; Lee, B.J.; Kim, K.I.; Jung, H.J.; Jang, H.J. Rosa laevigata Attenuates Allergic Asthma Exacerbated by Water-Soluble PM by Downregulating the MAPK Pathway. Front. Pharmacol. 2022, 13, 925502. [Google Scholar] [CrossRef] [PubMed]
- Grünenwald, M.; Adams, M.B.; Carter, C.G.; Nichols, D.S.; Koppe, W.; Verlhac-Trichet, V.; Schierle, J.; Adams, L.R. Pigment-depletion in Atlantic salmon (Salmo salar) post-smolt starved at elevated temperature is not influenced by dietary carotenoid type and increasing α-tocopherol level. Food Chem. 2019, 299, 125140. [Google Scholar] [CrossRef]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. α-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Hu, M.; Yang, J.; Xu, Y. Effect of α-tocopherol in alleviating the lipopolysaccharide-induced acute lung injury via inhibiting nuclear factor kappa-B signaling pathways. Bioengineered 2022, 13, 3958–3968. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, C.; Zou, N.; Chen, Q.; Wang, C.; Zhou, X.; Luo, L.; Qi, H.; Li, J.; Liu, Z.; et al. Lipocalin-2 silencing suppresses inflammation and oxidative stress of acute respiratory distress syndrome by ferroptosis via inhibition of MAPK/ERK pathway in neonatal mice. Bioengineered 2022, 13, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.G.; Chang, C.Y.; Yen, C.Y.; Lai, C.C. Associations between environmental heavy metal exposure and childhood asthma: A population-based study. J. Microbiol. Immunol. Infect. 2019, 52, 352–362. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Hu, X.; Jiang, Y.; Tang, J.; Ge, H.; Deng, S.; Li, X.; Feng, J. Involvement of ERK and Oxidative Stress in Airway Exposure to Cadmium Chloride Aggravates Airway Inflammation in Ovalbumin-Induced Asthmatic Mice. Toxics 2024, 12, 235. https://doi.org/10.3390/toxics12040235
Wu C, Hu X, Jiang Y, Tang J, Ge H, Deng S, Li X, Feng J. Involvement of ERK and Oxidative Stress in Airway Exposure to Cadmium Chloride Aggravates Airway Inflammation in Ovalbumin-Induced Asthmatic Mice. Toxics. 2024; 12(4):235. https://doi.org/10.3390/toxics12040235
Chicago/Turabian StyleWu, Chendong, Xinyue Hu, Yuanyuan Jiang, Jiale Tang, Huan Ge, Shuanglinzi Deng, Xiaozhao Li, and Juntao Feng. 2024. "Involvement of ERK and Oxidative Stress in Airway Exposure to Cadmium Chloride Aggravates Airway Inflammation in Ovalbumin-Induced Asthmatic Mice" Toxics 12, no. 4: 235. https://doi.org/10.3390/toxics12040235
APA StyleWu, C., Hu, X., Jiang, Y., Tang, J., Ge, H., Deng, S., Li, X., & Feng, J. (2024). Involvement of ERK and Oxidative Stress in Airway Exposure to Cadmium Chloride Aggravates Airway Inflammation in Ovalbumin-Induced Asthmatic Mice. Toxics, 12(4), 235. https://doi.org/10.3390/toxics12040235