
Cadmium Exposure: Mechanisms and Pathways of Toxicity and Implications for Human Health

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Absorption, Distribution, Metabolism, and Excretion (ADME) Process of Cadmium in the Body
3. Oxidative Stress Caused by Cadmium
4. Cadmium Impact on Signal Transduction Pathways
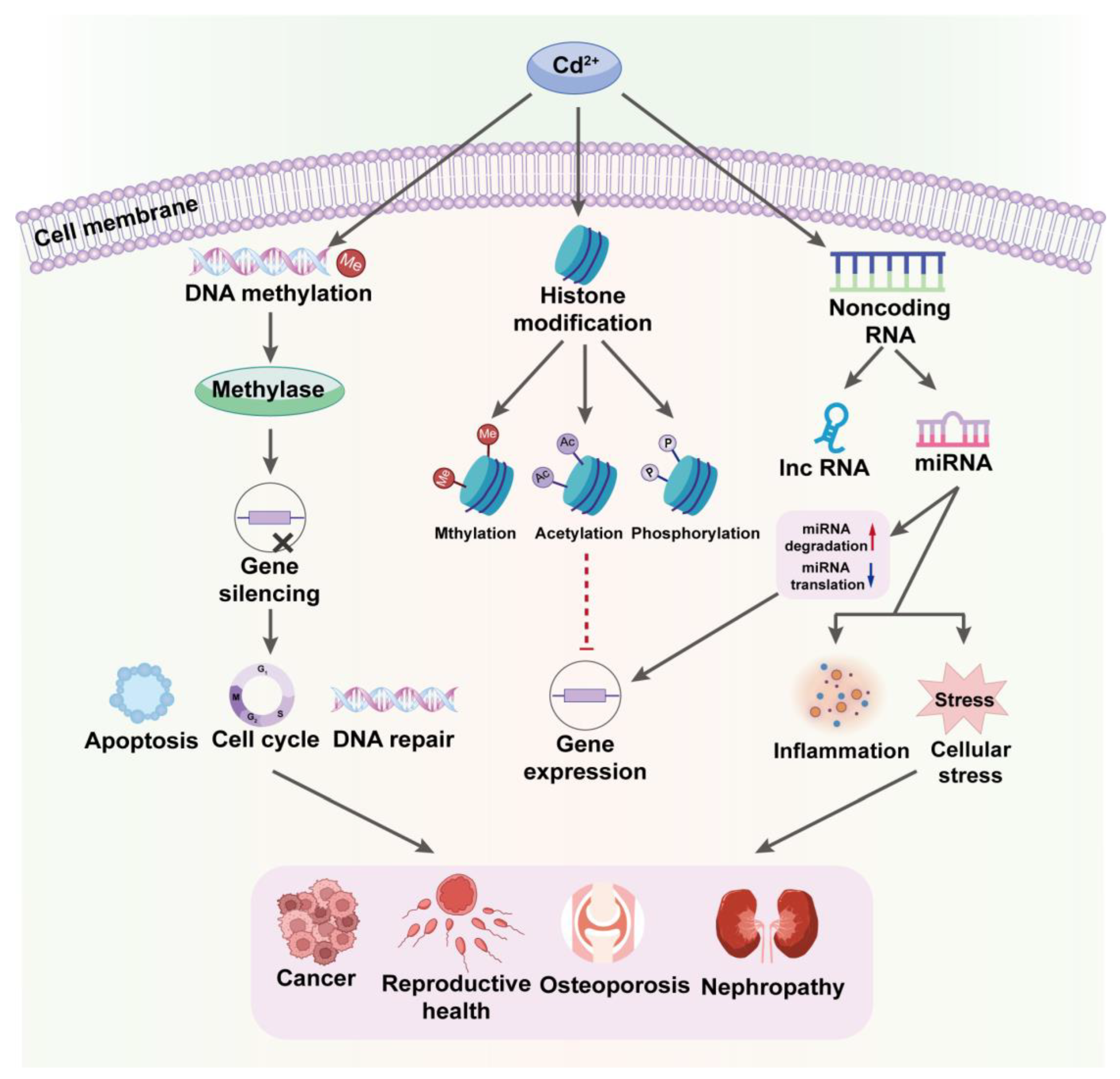
5. Cadmium-Induced Epigenetic Alterations
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayat, M.T.; Nauman, M.; Nazir, N.; Ali, S.; Bangash, N. Environmental hazards of cadmium: Past, present, and future. In Cadmium Toxicity and Tolerance in Plants; Elsevier: Amsterdam, The Netherlands, 2019; pp. 163–183. [Google Scholar]
- Wang, M.; Chen, Z.; Song, W.; Hong, D.; Huang, L.; Li, Y. A review on cadmium exposure in the population and intervention strategies against cadmium toxicity. Bull. Environ. Contam. Toxicol. 2021, 106, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, T.S.; Staessen, J.A.; Roels, H.A.; Munters, E.; Cuypers, A.; Richart, T.; Ruttens, A.; Smeets, K.; Clijsters, H.; Vangronsveld, J. Cadmium exposure in the population: From health risks to strategies of prevention. Biometals 2010, 23, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Elinder, C.-G. Cadmium: Uses, occurrence, and intake. In Cadmium and Health; CRC Press: Boca Raton, FL, USA, 2019; pp. 23–64. [Google Scholar]
- Rietra, R.; Mol, G.; Rietjens, I.; Romkens, P. Cadmium in Soil, Crops and Resultant Dietary Exposure; Wageningen Environmental Research: Wageningen, The Netherlands, 2017. [Google Scholar]
- Shakya, P.R.; Shrestha, P.; Tamrakar, C.S.; Bhattarai, P.K. Studies and determination of heavy metals in waste tyres and their impacts on the environment. Pak. J. Anal. Environ. Chem. 2006, 7, 7. [Google Scholar]
- Kim, H.; Lee, H.J.; Hwang, J.-Y.; Ha, E.-H.; Park, H.; Ha, M.; Kim, J.H.; Hong, Y.-C.; Chang, N. Blood cadmium concentrations of male cigarette smokers are inversely associated with fruit consumption. J. Nutr. 2010, 140, 1133–1138. [Google Scholar] [CrossRef]
- Satarug, S.; Haswell-Elkins, M.R.; Moore, M.R. Safe levels of cadmium intake to prevent renal toxicity in human subjects. Br. J. Nutr. 2000, 84, 791–802. [Google Scholar] [CrossRef]
- Charkiewicz, A.E.; Omeljaniuk, W.J.; Nowak, K.; Garley, M.; Nikliński, J. Cadmium Toxicity and Health Effects—A Brief Summary. Molecules 2023, 28, 6620. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Wilson, A.; Rajan, S.; Jonah, M. Biochemical pathways in cadmium toxicity. Mol. Biol. Toxicol. Met. 2000, 34–74. [Google Scholar]
- Sabolić, I.; Breljak, D.; Škarica, M.; Herak-Kramberger, C.M. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 2010, 23, 897–926. [Google Scholar] [CrossRef]
- Tomza-Marciniak, A.; Pilarczyk, B.; Marciniak, A.; Udała, J.; Bąkowska, M.; Pilarczyk, R. Cadmium, Cd. In Mammals and Birds as Bioindicators of Trace Element Contaminations in Terrestrial Environments: An Ecotoxicological Assessment of the Northern Hemisphere; Springer: Berlin/Heidelberg, Germany, 2019; pp. 483–532. [Google Scholar]
- Prozialeck, W.C.; Edwards, J.R. Early biomarkers of cadmium exposure and nephrotoxicity. Biometals 2010, 23, 793–809. [Google Scholar] [CrossRef]
- Gerhardsson, L.; Skerfving, S. Concepts on biological markers and biomonitoring for metal toxicity. In Toxicology of Metals, Volume I; CRC Press: Boca Raton, FL, USA, 2023; pp. 81–107. [Google Scholar]
- Bernard, A. Renal dysfunction induced by cadmium: Biomarkers of critical effects. Biometals 2004, 17, 519–523. [Google Scholar] [CrossRef]
- Huang, M.; Choi, S.J.; Kim, D.W.; Kim, N.Y.; Bae, H.S.; Yu, S.D.; Kim, D.S.; Kim, H.; Choi, B.S.; Yu, I.J. Evaluation of factors associated with cadmium exposure and kidney function in the general population. Environ. Toxicol. 2013, 28, 563–570. [Google Scholar] [CrossRef]
- Noonan, C.W.; Sarasua, S.M.; Campagna, D.; Kathman, S.J.; Lybarger, J.A.; Mueller, P.W. Effects of exposure to low levels of environmental cadmium on renal biomarkers. Environ. Health Perspect. 2002, 110, 151–155. [Google Scholar] [CrossRef]
- Niede, R.; Benbi, D.K. Integrated review of the nexus between toxic elements in the environment and human health. AIMS Public Health 2022, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic metals and oxidative stress part I: Mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem. 2001, 1, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef]
- Nies, D.H.; Grass, G. Transition metal homeostasis. EcoSal Plus 2009, 3. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Mohammed, M.T.; Kadhim, S.M.; Jassimand, A.; Abbas, S. Free radicals and human health. Int. J. Innov. Sci. Res. 2015, 4, 218–223. [Google Scholar]
- Branca, J.J.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-induced oxidative stress: Focus on the central nervous system. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 2010, 198, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cruz, E.Y.; Amador-Martínez, I.; Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Chaverri, J.P. Renal damage induced by cadmium and its possible therapy by mitochondrial transplantation. Chem. Biol. Interact. 2022, 361, 109961. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Green, D.R. Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 1999, 274, 17484–17490. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2017, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Das, S.C.; Al-Naemi, H.A. Cadmium Toxicity: Oxidative Stress, Inflammation and Tissue Injury. Occup. Dis. Environ. Med. 2019, 4, 144–163. [Google Scholar] [CrossRef]
- Hossein-Khannazer, N.; Azizi, G.; Eslami, S.; Alhassan Mohammed, H.; Fayyaz, F.; Hosseinzadeh, R.; Usman, A.B.; Kamali, A.N.; Mohammadi, H.; Jadidi-Niaragh, F. The effects of cadmium exposure in the induction of inflammation. Immunopharmacol. Immunotoxicol. 2020, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Nichols, L.; Dave, A.A.; Pittman, E.H.; Cheek, J.P.; Caroland, A.J.; Lotwala, P.; Drummond, J.; Bridges, C.C. Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 11105. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Stadtman, E.; Levine, R. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef]
- Li, J.; O, W.; Li, W.; Jiang, Z.-G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S. Reactive oxygen species and cellular defense system. Free. Radic. Hum. Health Dis. 2015, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Sun, J.; Wu, B.; Yuan, Y.; Gu, J.; Bian, J.; Liu, X.; Liu, Z. Effects of cadmium and/or lead on autophagy and liver injury in rats. Biol. Trace Elem. Res. 2020, 198, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cruz, E.Y.; Arancibia-Hernández, Y.L.; Loyola-Mondragón, D.Y.; Pedraza-Chaverri, J. Oxidative stress and its role in Cd-induced epigenetic modifications: Use of antioxidants as a possible preventive strategy. Oxygen 2022, 2, 177–210. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Waisberg, M.; Joseph, P.; Hale, B.; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar] [CrossRef]
- Templeton, D.M.; Liu, Y. Multiple roles of cadmium in cell death and survival. Chem. Biol. Interact. 2010, 188, 267–275. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Shelton, J.G.; Lee, J.T.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.; McCubrey, J.A. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway. Int. J. Oncol. 2003, 22, 469–480. [Google Scholar]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Su, Q.; Yue, C.; Zou, H.; Zhu, J.; Zhao, H.; Song, R.; Liu, Z. The effect of oxidative stress-induced autophagy by cadmium exposure in kidney, liver, and bone damage, and neurotoxicity. Int. J. Mol. Sci. 2022, 23, 13491. [Google Scholar] [CrossRef] [PubMed]
- Zanke, B.W.; Boudreau, K.; Rubie, E.; Winnett, E.; Tibbles, L.A.; Zon, L.; Kyriakis, J.; Liu, F.-F.; Woodgett, J.R. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr. Biol. 1996, 6, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F. Cadmium and cellular signaling cascades: To be or not to be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Arch. Toxicol. 2008, 82, 273–299. [Google Scholar] [CrossRef]
- Chuang, S.-M.; Wang, I.-C.; Yang, J.-L. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 2000, 21, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Benoit, B.; Baillet, A.; Poüs, C. Cytoskeleton and associated proteins: Pleiotropic JNK substrates and regulators. Int. J. Mol. Sci. 2021, 22, 8375. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Cheng, Z.; Xu, Y.; Wang, X.; Li, X.; Xu, D.; Kapron, C.M.; Liu, J. Low dose cadmium inhibits proliferation of human renal mesangial cells via activation of the JNK pathway. Int. J. Environ. Res. Public Health 2016, 13, 990. [Google Scholar] [CrossRef]
- Sarkar, A.; Ravindran, G.; Krishnamurthy, V. A brief review on the effect of cadmium toxicity: From cellular to organ level. Int. J. Biotechnol. Res. 2013, 3, 17–36. [Google Scholar]
- Kefaloyianni, E.; Gourgou, E.; Ferle, V.; Kotsakis, E.; Gaitanaki, C.; Beis, I. Acute thermal stress and various heavy metals induce tissue-specific pro-or anti-apoptotic events via the p38-MAPK signal transduction pathway in Mytilus galloprovincialis (Lam.). J. Exp. Biol. 2005, 208, 4427–4436. [Google Scholar] [CrossRef] [PubMed]
- Nemmiche, S. Oxidative signaling response to cadmium exposure. Toxicol. Sci. 2017, 156, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Mitra, S.; Chakraborty, A.J.; Tareq, A.M.; Emran, T.B.; Nainu, F.; Khusro, A.; Idris, A.M.; Khandaker, M.U.; Osman, H.; Alhumaydhi, F.A. Impact of heavy metals on the environment and human health: Novel therapeutic insights to counter the toxicity. J. King Saud Univ. Sci. 2022, 34, 101865. [Google Scholar] [CrossRef]
- Olszowski, T.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Pro-inflammatory properties of cadmium. Acta Biochim. Pol. 2012, 59, 475–482. [Google Scholar] [CrossRef]
- Sun, Z.; Andersson, R. NF-κB activation and inhibition: A review. Shock 2002, 18, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.-K. Cadmium and cellular signaling cascades: Interactions between cell death and survival pathways. Arch. Toxicol. 2013, 87, 1743–1786. [Google Scholar] [CrossRef] [PubMed]
- Sepand, M.-R.; Aliomrani, M.; Hasani-Nourian, Y.; Khalhori, M.-R.; Farzaei, M.-H.; Sanadgol, N. Mechanisms and pathogenesis underlying environmental chemical-induced necroptosis. Environ. Sci. Pollut. Res. 2020, 27, 37488–37501. [Google Scholar] [CrossRef] [PubMed]
- Đukić-Ćosić, D.; Baralić, K.; Javorac, D.; Djordjevic, A.B.; Bulat, Z. An overview of molecular mechanisms in cadmium toxicity. Curr. Opin. Toxicol. 2020, 19, 56–62. [Google Scholar] [CrossRef]
- Freitas, M.; Fernandes, E. Zinc, cadmium and nickel increase the activation of NF-κB and the release of cytokines from THP-1 monocytic cells. Metallomics 2011, 3, 1238–1243. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Méplan, C.; Mann, K.; Hainaut, P. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J. Biol. Chem. 1999, 274, 31663–31670. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Filipič, M. Mechanisms of cadmium induced genomic instability. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2012, 733, 69–77. [Google Scholar] [CrossRef]
- Amaral, J.D.; Castro, R.E.; Steer, C.J.; Rodrigues, C.M. p53 and the regulation of hepatocyte apoptosis: Implications for disease pathogenesis. Trends Mol. Med. 2009, 15, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K.; Paules, R.S. DNA damage and cell cycle checkpoints. FASEB J. 1996, 10, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Giorgi, C.; Romagnoli, A.; Pinton, P. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar]
- Gerzen, O.P.; Votinova, V.O.; Potoskueva, I.K.; Tzybina, A.E.; Nikitina, L.V. Direct Effects of Toxic Divalent Cations on Contractile Proteins with Implications for the Heart: Unraveling Mechanisms of Dysfunction. Int. J. Mol. Sci. 2023, 24, 10579. [Google Scholar] [CrossRef]
- Yang, W.Y.; Zhang, Z.Y.; Thijs, L.; Cauwenberghs, N.; Wei, F.F.; Jacobs, L.; Luttun, A.; Verhamme, P.; Kuznetsova, T.; Nawrot, T.S. Left ventricular structure and function in relation to environmental exposure to lead and cadmium. J. Am. Heart Assoc. 2017, 6, e004692. [Google Scholar] [CrossRef]
- Vangheluwe, P.; Sepulveda, M.R.; Missiaen, L.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. Intracellular Ca2+-and Mn2+-transport ATPases. Chem. Rev. 2009, 109, 4733–4759. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Liu, N.; Wang, Q.; Luo, J.; Wang, L. Cadmium induces apoptosis in freshwater crab Sinopotamon henanense through activating calcium signal transduction pathway. PLoS ONE 2015, 10, e0144392. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Y.; Xu, B.; Guo, M.; Zhang, Z.; Liu, L.; Ma, H.; Chen, Z.; Luo, Y.; Huang, S. CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J. Neurochem. 2011, 119, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xu, C.; Ran, D.; Wang, Y.; Zhao, H.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. CaMKII mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology 2018, 406, 70–80. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y.; Shao, C.; Tan, Y.; Cai, L. Cadmium and its epigenetic effects. Curr. Med. Chem. 2012, 19, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Venza, M.; Visalli, M.; Biondo, C.; Oteri, R.; Agliano, F.; Morabito, S.; Caruso, G.; Caffo, M.; Teti, D.; Venza, I. Epigenetic effects of cadmium in cancer: Focus on melanoma. Curr. Genom. 2014, 15, 420–435. [Google Scholar] [CrossRef]
- Zimta, A.-A.; Schitcu, V.; Gurzau, E.; Stavaru, C.; Manda, G.; Szedlacsek, S.; Berindan-Neagoe, I. Biological and molecular modifications induced by cadmium and arsenic during breast and prostate cancer development. Environ. Res. 2019, 178, 108700. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Freeman, J.L.; Xie, J.; Zhao, H. The Role of Dynamic Epigenetic Changes in Modulating Homeostasis after Exposure to Low-dose Environmental Chemicals. Genom. Epigenom. Biomark. Toxicol. Dis. Clin. Ther. Actions 2022, 213–228. [Google Scholar] [CrossRef]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The effects of cadmium toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Tariba Lovaković, B.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M. Toxic-metal-induced alteration in miRNA expression profile as a proposed mechanism for disease development. Cells 2020, 9, 901. [Google Scholar] [CrossRef]
- Martínez-Pacheco, M.; Hidalgo-Miranda, A.; Romero-Córdoba, S.; Valverde, M.; Rojas, E. MRNA and miRNA expression patterns associated to pathways linked to metal mixture health effects. Gene 2014, 533, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Anetor, J.I. Rising environmental cadmium levels in developing countries: Threat to genome stability and health. Niger. J. Physiol. Sci. 2012, 27, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, A. Cadmium toxicity: Effects on human reproduction and fertility. Rev. Environ. Health 2019, 34, 327–338. [Google Scholar] [CrossRef]
- De Angelis, C.; Galdiero, M.; Pivonello, C.; Salzano, C.; Gianfrilli, D.; Piscitelli, P.; Lenzi, A.; Colao, A.; Pivonello, R. The environment and male reproduction: The effect of cadmium exposure on reproductive function and its implication in fertility. Reprod. Toxicol. 2017, 73, 105–127. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, F.; Zheng, W. Cadmium Exposure: Mechanisms and Pathways of Toxicity and Implications for Human Health. Toxics 2024, 12, 388. https://doi.org/10.3390/toxics12060388
Qu F, Zheng W. Cadmium Exposure: Mechanisms and Pathways of Toxicity and Implications for Human Health. Toxics. 2024; 12(6):388. https://doi.org/10.3390/toxics12060388
Chicago/Turabian StyleQu, Fei, and Weiwei Zheng. 2024. "Cadmium Exposure: Mechanisms and Pathways of Toxicity and Implications for Human Health" Toxics 12, no. 6: 388. https://doi.org/10.3390/toxics12060388