
Effects on Iron Metabolism and System


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture and Reagents
2.2. Cell Viability Assays
2.3. Determination of Intracellular Content
2.4. Detection of Intracellular GSH and Malondialdehyde (MDA) Levels
2.5. Detection of Intracellular Lipid ROS Content
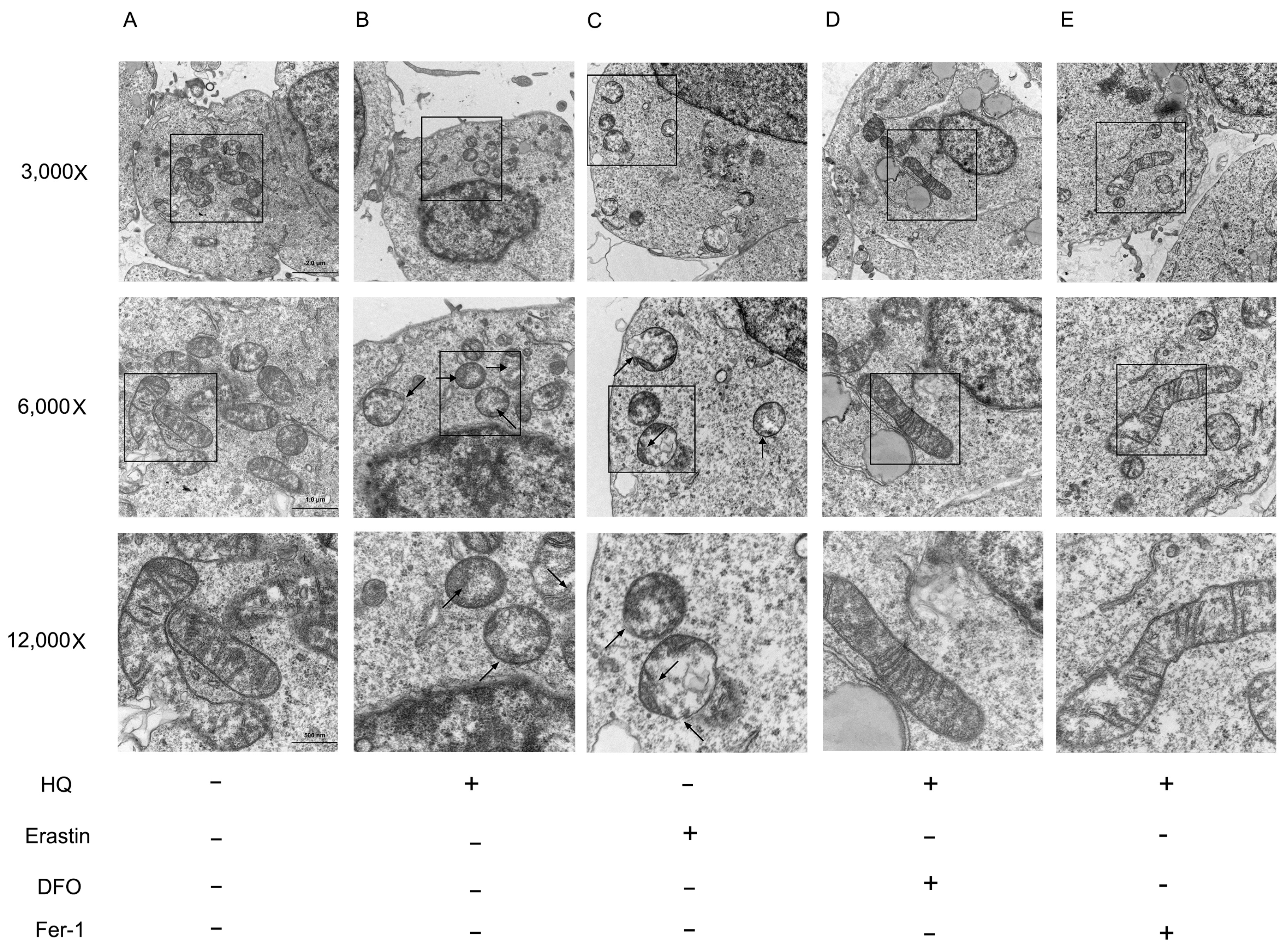
2.6. Observation of Mitochondrial Ultrastructure
2.7. Total RNA Extraction, Reverse Transcription and Quantitative Real-Time PCR
2.8. Protein Extraction and Western Blot Analysis
2.9. Statistical Analysis
3. Results
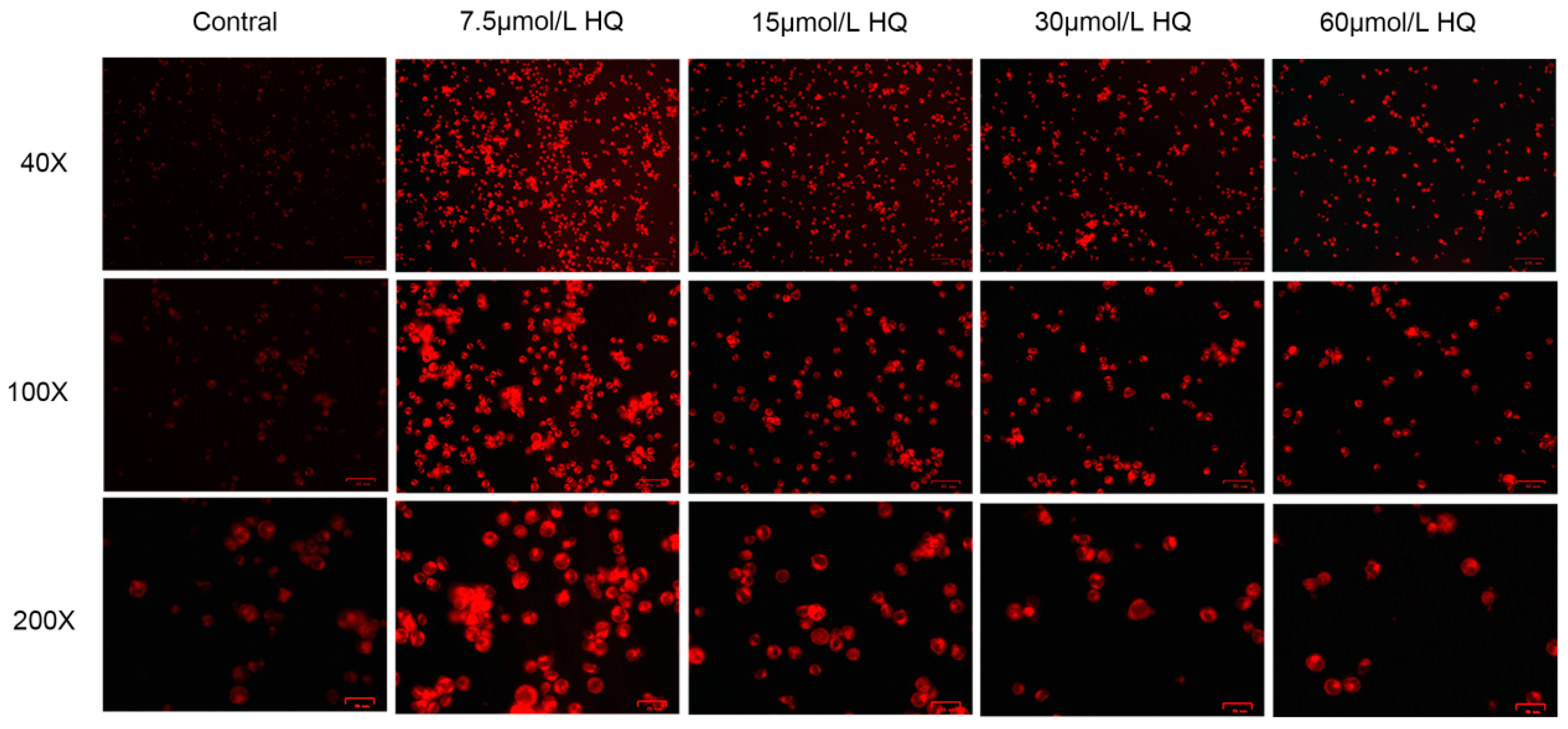
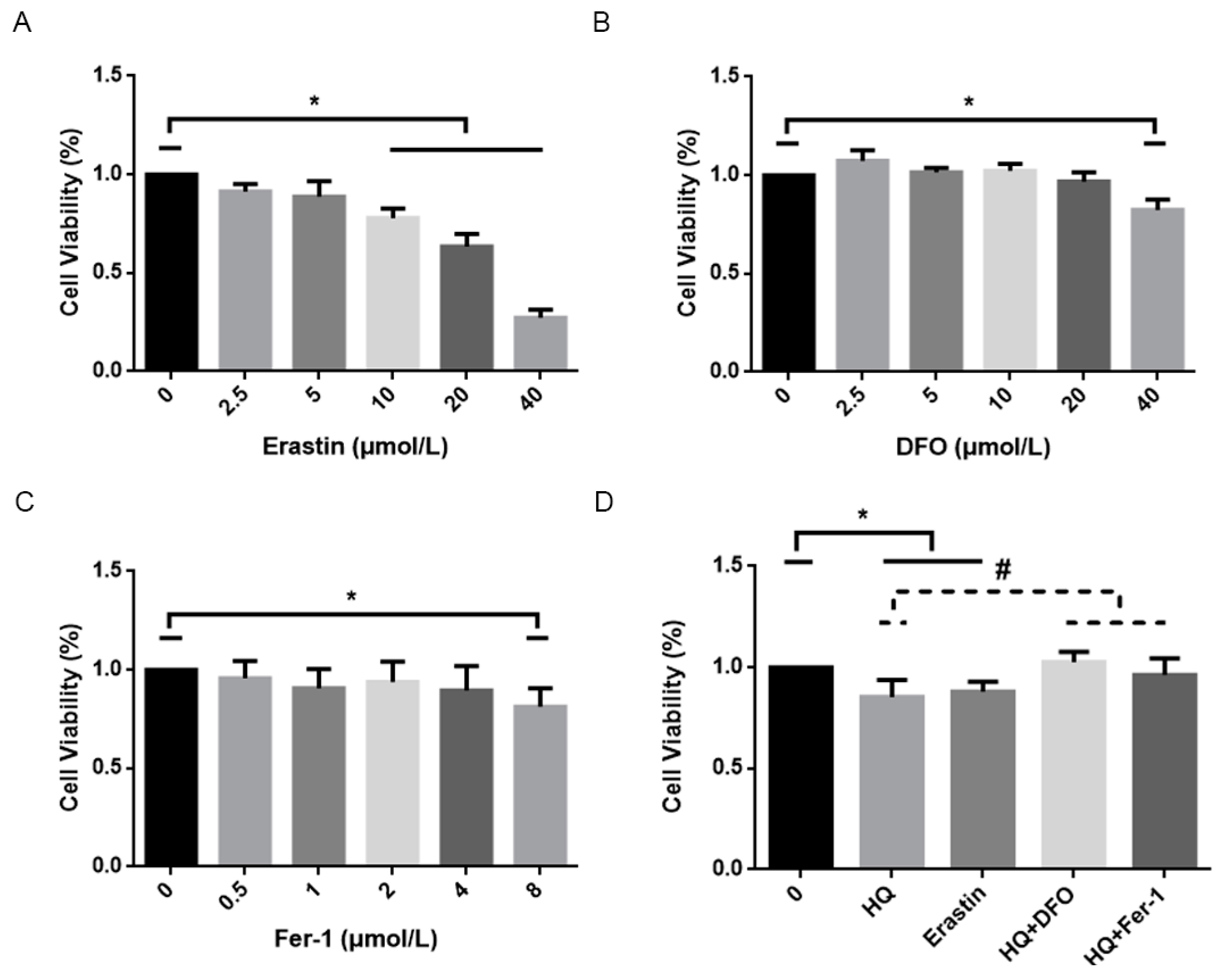
3.1. HQ Has a Significant Toxic Effect on Jurkat Cells
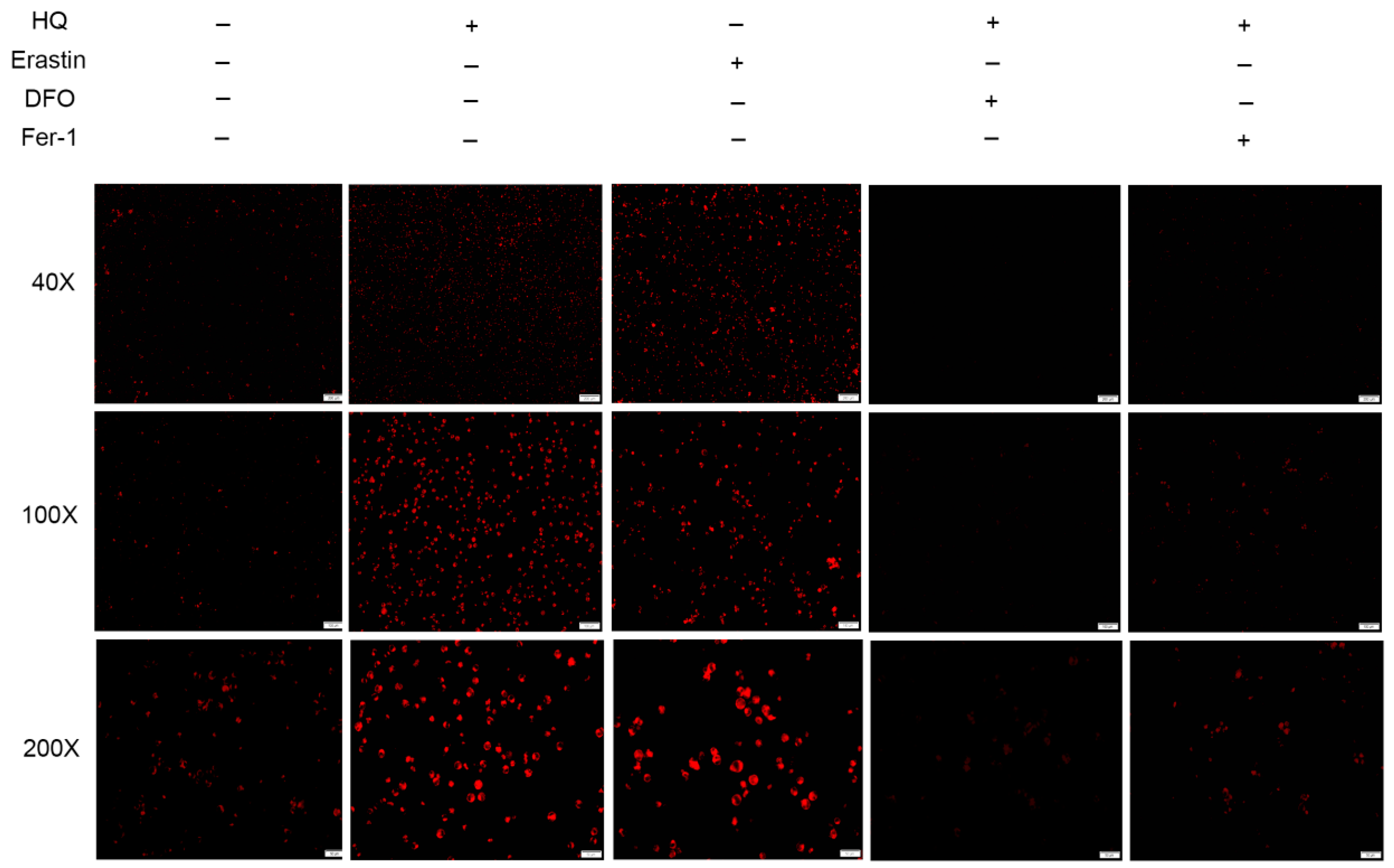
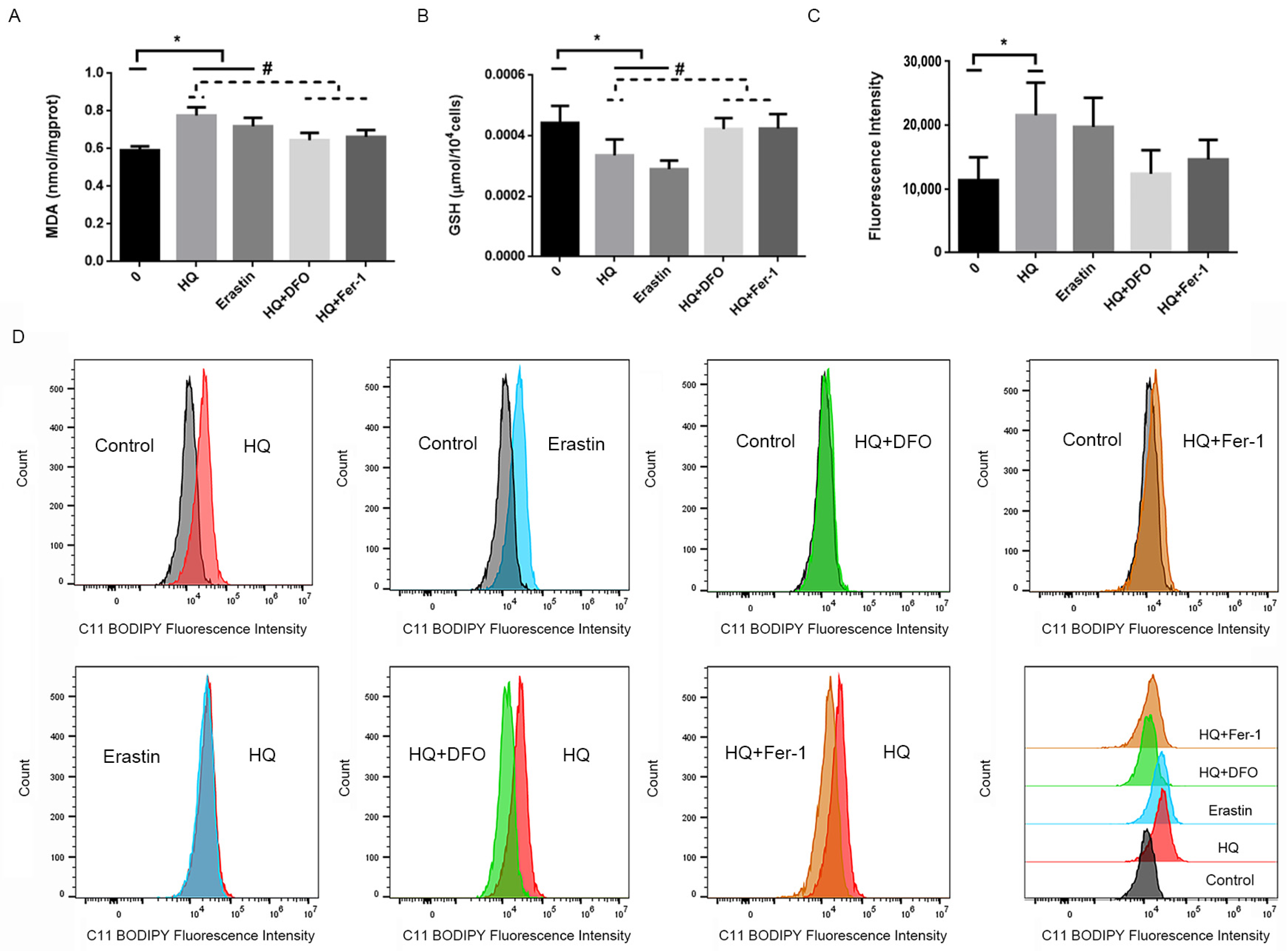
3.2. HQ Induces Ferroptosis in Jurkat Cells
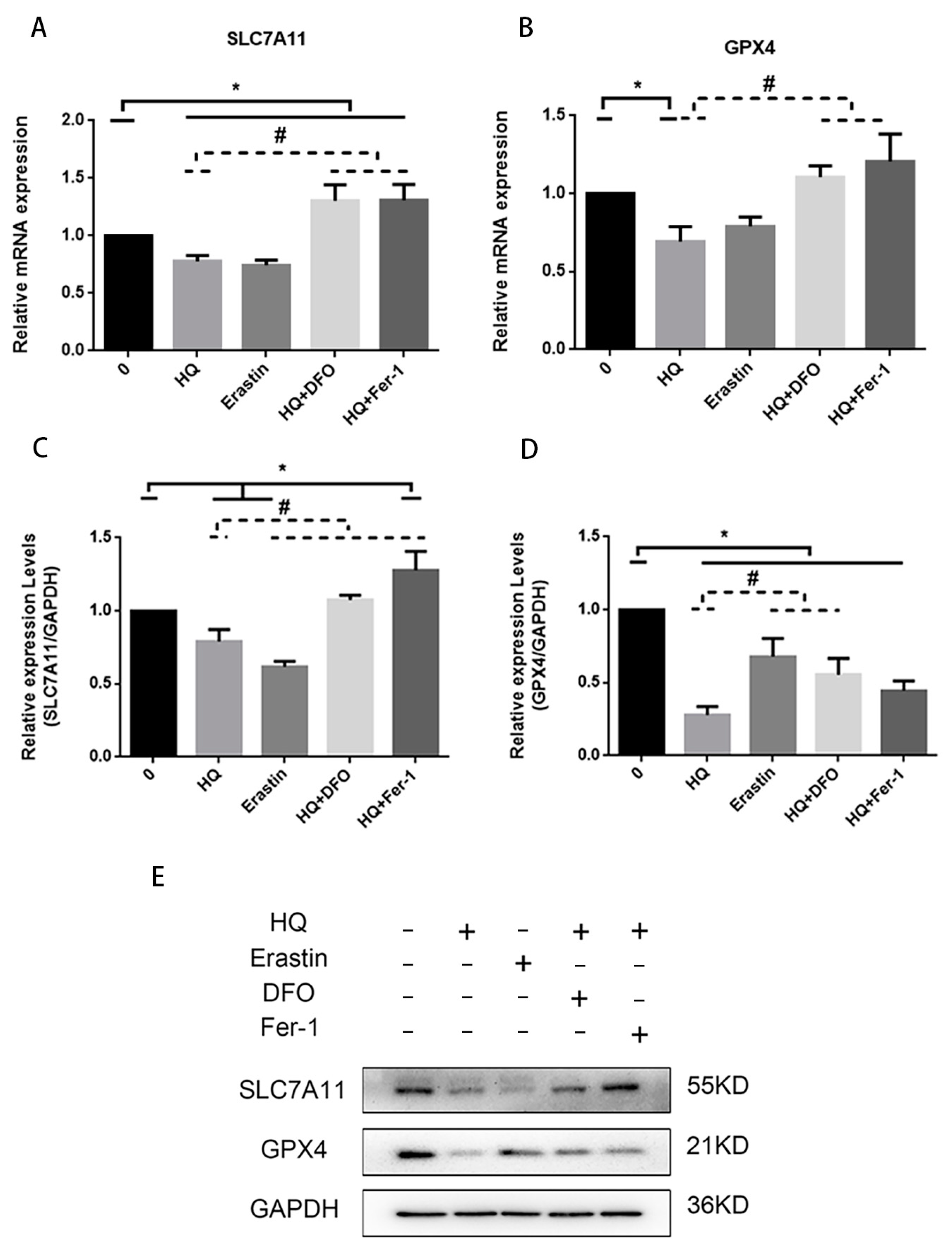
3.3. Iron Metabolism Pathway and System /GPX4 Pathway Involved in HQ-Induced Ferroptosis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aksoy, M.; Erdem, S.; DinCol, G. Leukemia in shoe-workers exposed chronically to benzene. Blood 1974, 44, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Health 2010, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, B.; Xu, S.; Cong, X.; Pu, Y.; Zhang, J. Research development and trends of benzene-induced leukemia from 1990 to 2019-A bibliometric analysis. Environ. Sci. Pollut. Res. Int. 2022, 29, 9626–9639. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.L.; Kalf, G.F. Induced differentiation of HL-60 promyelocytic leukemia cells to monocyte/macrophages is inhibited by hydroquinone, a hematotoxic metabolite of benzene. Blood 1992, 79, 627–633. [Google Scholar] [CrossRef]
- Qian, S.; Han, Y.; Shi, Y.; Xu, W.; Zhu, Y.; Jiang, S.; Chen, Y.; Yu, Z.; Zhang, S.; Yang, Y.; et al. Benzene induces haematotoxicity by promoting deacetylation and autophagy. J. Cell Mol. Med. 2019, 23, 1022–1033. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, H.; Pan, Z.; Ling, X.; Wu, M.; Gui, Z.; Chen, J.; Peng, J.; Liu, Z.; Tan, Q.; et al. Regulatory loop between lncRNA FAS-AS1 and DNMT3b controls FAS expression in hydroquinone-treated TK6 cells and benzene-exposed workers. Environ. Pollut. 2020, 261, 114147. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Xi, C.; Pang, J.; Zhi, W.; Chang, C.S.; Siddaramappa, U.; Shi, H.; Horuzsko, A.; Pace, B.S.; Zhu, X. Nrf2 sensitizes ferroptosis through l-2-hydroxyglutarate-mediated chromatin modifications in sickle cell disease. Blood 2023, 142, 382–396. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, H.; Zhang, W.; Wang, J.; Liu, Z.; Jing, J.; Han, L.; Gao, A. Probiotics ameliorate benzene-induced systemic inflammation and hematopoietic toxicity by inhibiting Bacteroidaceae-mediated ferroptosis. Sci. Total Environ. 2023, 899, 165678. [Google Scholar] [CrossRef]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Liggett, L.A.; Sankaran, V.G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182, 1384–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jia, Y.; Mahmut, D.; Deik, A.A.; Jeanfavre, S.; Clish, C.B.; Sankaran, V.G. Human hematopoietic stem cell vulnerability to ferroptosis. Cell 2023, 186, 732–747. [Google Scholar] [CrossRef]
- Bhattarai, N.; Korhonen, E.; Mysore, Y.; Kaarniranta, K.; Kauppinen, A. Hydroquinone induces NLRP3-independent IL-18 release from ARPE-19 cells. Cells 2021, 10, 1405. [Google Scholar] [CrossRef]
- Bhattarai, N.; Korhonen, E.; Toppila, M.; Koskela, A.; Kaarniranta, K.; Mysore, Y.; Kauppinen, A. Resvega alleviates hydroquinone-induced oxidative stress in ARPE-19 cells. Int. J. Mol. Sci. 2020, 21, 2066. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Nakamura, T.; Motooka, Y.; Ito, F.; Jiang, L.; Akatsuka, S.; Iwase, A.; Kajiyama, H.; Kikkawa, F.; Toyokuni, S. Novel ovarian endometriosis model causes infertility via iron-mediated oxidative stress in mice. Redox Biol. 2020, 37, 101726. [Google Scholar] [CrossRef]
- Wang, W.; Jing, X.; Du, T.; Ren, J.; Liu, X.; Chen, F.; Shao, Y.; Sun, S.; Yang, G.; Cui, X. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic. Biol. Med. 2022, 190, 234–246. [Google Scholar] [CrossRef]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Pei, Z.; Qin, Y.; Fu, X.; Yang, F.; Huo, F.; Liang, X.; Wang, S.; Cui, H.; Lin, P.; Zhou, G.; et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. 2022, 57, 102509. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Chen, X.; Feng, J.; Duan, T.; Liu, S.; Sun, X.; Chen, P.; Pan, T.; Yan, L.; Jin, T.; et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm. Sin. B 2021, 11, 4045–4054. [Google Scholar] [CrossRef]
- Peng, C.; Arthur, D.; Liu, F.; Lee, J.; Xia, Q.; Lavin, M.F.; Ng, J.C. Genotoxicity of hydroquinone in A549 cells. Cell Biol. Toxicol. 2013, 29, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chou, W.M.; Chang, L.S. p38 MAPK/PP2Acalpha/TTP pathway on the connection of TNF-alpha and caspases activation on hydroquinone-induced apoptosis. Carcinogenesis 2013, 34, 818–827. [Google Scholar] [CrossRef]
- Chen, L.; Zhai, L.; Gao, Y.; Cui, Z.; Yu, L.; Zhu, D.; Tang, H.; Luo, H. Nrf2 affects hydroquinone-induces cell cycle arrest through the p16/pRb signaling pathway and antioxidant enzymes. Ecotoxicol. Environ. Saf. 2023, 249, 114389. [Google Scholar] [CrossRef]
- Inayat-Hussain, S.H.; Winski, S.L.; Ross, D. Differential involvement of caspases in hydroquinone-induced apoptosis in human leukemic hl-60 and jurkat cells. Toxicol. Appl. Pharmacol. 2001, 175, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Lee, Y.C.; Chiou, J.T.; Chen, Y.J.; Chang, L.S. Effects of SIDT2 on the miR-25/NOX4/HuR axis and SIRT3 mRNA stability lead to ROS-mediated TNF-alpha expression in hydroquinone-treated leukemia cells. Cell Biol. Toxicol. 2023, 39, 2207–2225. [Google Scholar] [CrossRef]
- Yu, C.H.; Yang, S.Q.; Li, L.; Xin, Y.; Zhang, F.; Liu, X.F.; Yi, Z.C. Identification of potential pathways and microRNA-mRNA networks associated with benzene metabolite hydroquinone-induced hematotoxicity in human leukemia K562 cells. BMC Pharmacol. Toxicol. 2022, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxid. Med. Cell Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef] [PubMed]
- Dachert, J.; Schoeneberger, H.; Rohde, K.; Fulda, S. RSL3 and Erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget 2016, 7, 63779–63792. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, X.; Lu, D.; Yin, M.; Shan, M.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, M.; Nakatsuka, Y.; Vandenbon, A.; Ori, D.; Uehata, T.; Tsujimura, T.; Suzuki, Y.; Mino, T.; Takeuchi, O. Regnase-1 Maintains Iron Homeostasis via the Degradation of Transferrin Receptor 1 and Prolyl-Hydroxylase-Domain-Containing Protein 3 mRNAs. Cell Rep. 2017, 19, 1614–1630. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, T.; Xiang, Q.; Li, N.; Wang, J.; Liu, J.; Zhang, Y.; Yang, T.; Bian, J. GPR116 promotes ferroptosis in sepsis-induced liver injury by suppressing system Xc−/GSH/GPX4. Cell Biol. Toxicol. 2023, 39, 3015–3030. [Google Scholar] [CrossRef]
- He, Q.; Yang, J.; Pan, Z.; Zhang, G.; Chen, B.; Li, S.; Xiao, J.; Tan, F.; Wang, Z.; Chen, P.; et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System Xc−/GPX4 axis. Biomed. Pharmacother. 2023, 157, 113915. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Liu, G.; Li, Q.; Hu, Y.; Wang, H.
Effects on Iron Metabolism and System
Liu N, Liu G, Li Q, Hu Y, Wang H.
Effects on Iron Metabolism and System
Liu, Nana, Ge Liu, Qiang Li, Yipeng Hu, and Hong Wang.
2024. "Effects on Iron Metabolism and System
Liu, N., Liu, G., Li, Q., Hu, Y., & Wang, H.
(2024). Effects on Iron Metabolism and System