
Bisphenol S Induces Lipid Metabolism Disorders in HepG2 and SK-Hep-1 Cells via Oxidative Stress

Abstract
:1. Introduction
2. Experimental Method
2.1. Cell Culture and Exposure
2.2. CCK-8 Method Cell Viability Assays
2.3. ROS Reactive Oxygen Detection
2.4. Oil Red O Staining
2.5. Measurement of Biochemical Indicators and Oxidative Stress Indicators
2.6. Real-Time Quantitative PCR (qPCR) Analysis
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
3.1. Selection of the Best BPS Concentration
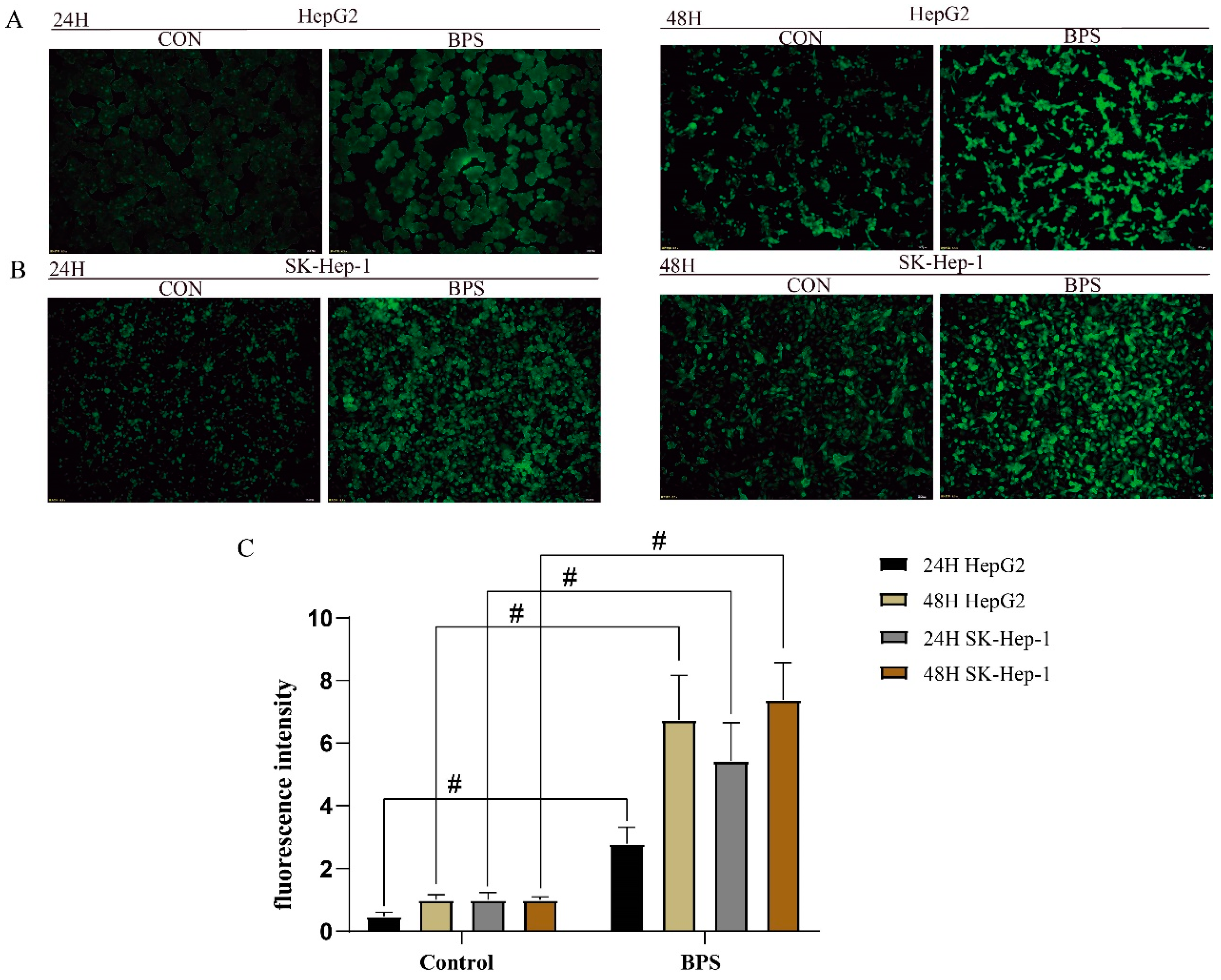
3.2. Effect of BPS on ROS Levels in HepG2 Cells and SK-Hep-1 Cells
3.3. BPS Induced Lipid Droplet Deposition in HepG2 and SK-Hep-1 Cells
3.4. Effect of BPS on MDA and CAT Levels in HepG2 and SK-Hep-1 Cells
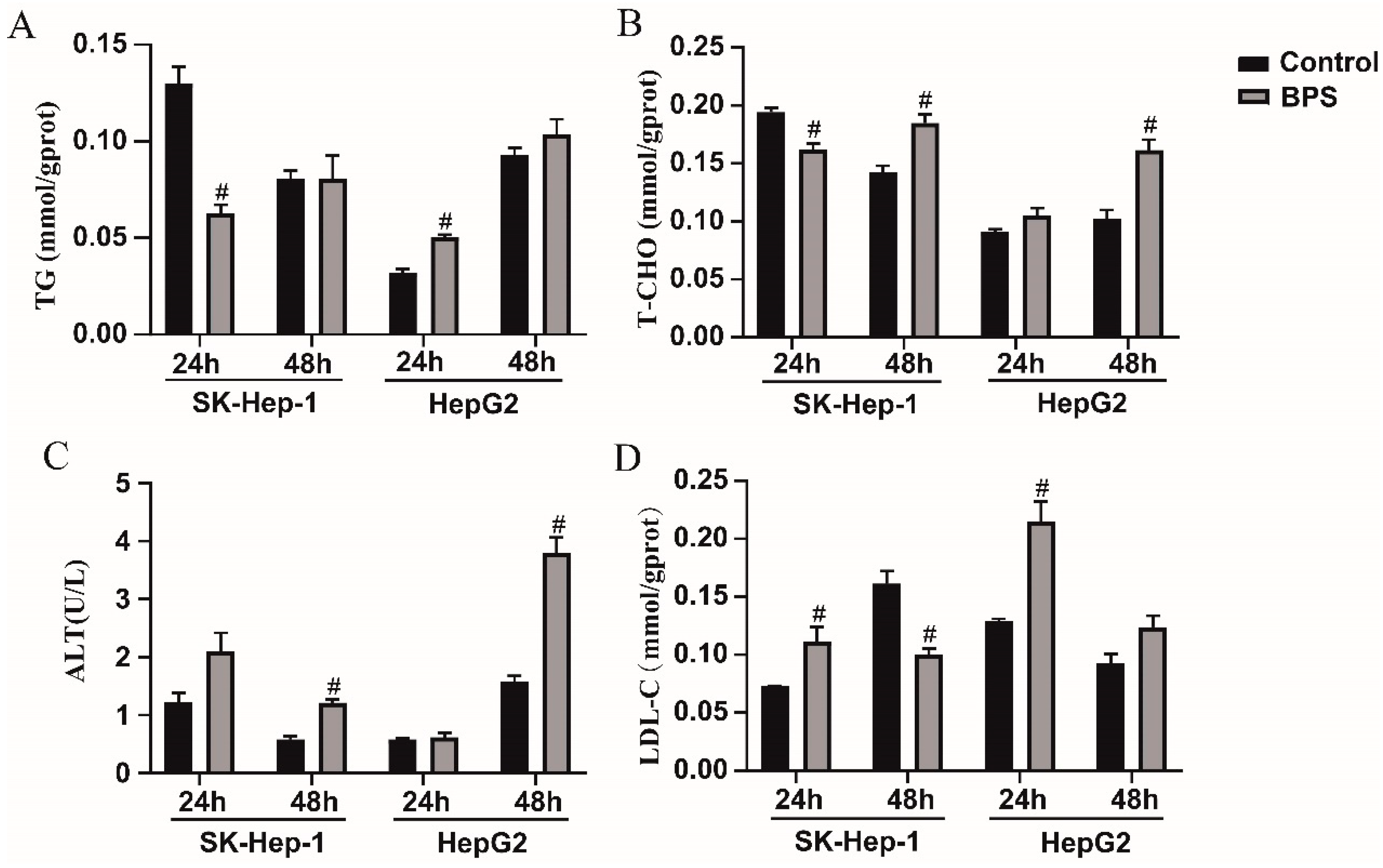
3.5. Effects of BPS on HepG2 and SK-Hep-1 Cell Damage and Metabolic-Related Indicators
3.6. Effects of BPS on PPARα, CPT1B, CD36, SREBP1C, and FASN mRNA Levels in HepG2 and SK-Hep-1 Cells
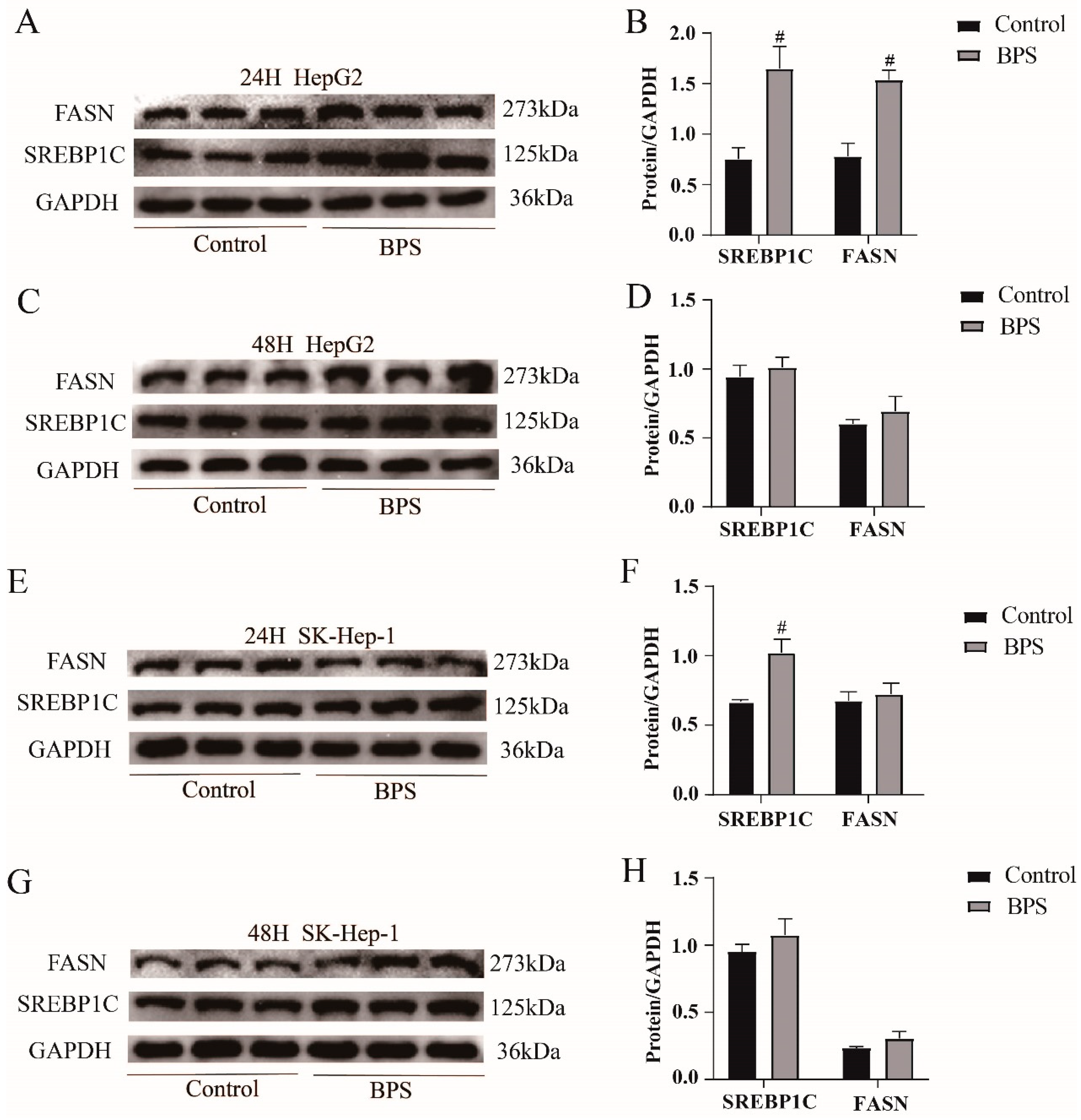
3.7. Effects of BPS on the Expression Levels of the Lipid Synthesis Proteins SREBP1C and FASN in HepG2 Cells and SK-Hep-1 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bousoumah, R.; Leso, V.; Iavicoli, I.; Huuskonen, P.; Viegas, S.; Porras, S.P.; Santonen, T.; Frery, N.; Robert, A.; Ndaw, S. Biomonitoring of occupational exposure to bisphenol A, bisphenol S and bisphenol F: A systematic review. Sci. Total Environ. 2021, 783, 146905. [Google Scholar] [CrossRef]
- Pu, C.; Liu, Y.; Zhu, J.; Ma, J.; Cui, M.; Mehdi, O.M.; Wang, B.; Wang, A.; Zhang, C. Mechanisms insights into bisphenol S-induced oxidative stress, lipid metabolism disruption, and autophagy dysfunction in freshwater crayfish. J. Hazard. Mater. 2024, 479, 135704. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Yu, H.; Wei, Y.; Liu, F.; Ye, J. Disrupted metabolic pathways and potential human diseases induced by bisphenol S. Environ. Toxicol. Pharmacol. 2021, 88, 103751. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, R.; Liang, W.; Wei, S.; Zhou, Y.; Zeng, F. Assessment of BPA and BPS exposure in the general population in Guangzhou, China-Estimation of daily intakes based on urinary metabolites. Environ. Pollut. 2022, 315, 120375. [Google Scholar] [CrossRef] [PubMed]
- Ndaw, S.; Remy, A.; Denis, F.; Marsan, P.; Jargot, D.; Robert, A. Occupational exposure of cashiers to bisphenol S via thermal paper. Toxicol. Lett. 2018, 298, 106–111. [Google Scholar] [CrossRef]
- Zhao, X.; Qiu, W.; Zheng, Y.; Xiong, J.; Gao, C.; Hu, S. Occurrence, distribution, bioaccumulation, and ecological risk of bisphenol analogues, parabens and their metabolites in the Pearl River Estuary, South China. Ecotoxicol. Environ. Saf. 2019, 180, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Danzl, E.; Sei, K.; Soda, S.; Ike, M.; Fujita, M. Biodegradation of bisphenol A, bisphenol F and bisphenol S in seawater. Int. J. Environ. Res. Public Health 2009, 6, 1472–1484. [Google Scholar] [CrossRef] [PubMed]
- Kahn, L.G.; Philippat, C.; Nakayama, S.F.; Slama, R.; Trasande, L. Endocrine-disrupting chemicals: Implications for human health. Lancet Diabetes Endocrinol. 2020, 8, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lehmler, H.J.; Sun, Y.; Xu, G.; Liu, Y.; Zong, G.; Sun, Q.; Hu, F.B.; Wallace, R.B.; Bao, W. Bisphenol A substitutes and obesity in US adults: Analysis of a population-based, cross-sectional study. Lancet Planet. Health 2017, 1, e114–e122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, T.; Hu, W.; Wang, X.; Xu, B.; Lin, Z.; Hofer, T.; Stefanoff, P.; Chen, Y.; Wang, X.; et al. Association between exposure to a mixture of phenols, pesticides, and phthalates and obesity: Comparison of three statistical models. Environ. Int. 2019, 123, 325–336. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, D.; Yan, S.; Li, R.; Yan, J.; Teng, M.; Zhou, Z.; Zhu, W. Effects of perinatal exposure to BPA and its alternatives (BPS, BPF and BPAF) on hepatic lipid and glucose homeostasis in female mice adolescent offspring. Chemosphere 2018, 212, 297–306. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, D.; Liu, W.; Li, R.; Yan, S.; Jia, M.; Zhang, L.; Zhou, Z.; Zhu, W. Perinatal exposure to Bisphenol S (BPS) promotes obesity development by interfering with lipid and glucose metabolism in male mouse offspring. Environ. Res. 2019, 173, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Mornagui, B.; Rezg, R.; Repond, C.; Pellerin, L. Bisphenol S favors hepatic steatosis development via an upregulation of liver MCT1 expression and an impairment of the mitochondrial respiratory system. J. Cell Physiol. 2022, 237, 3057–3068. [Google Scholar] [CrossRef]
- Gao, C.; He, H.; Qiu, W.; Zheng, Y.; Chen, Y.; Hu, S.; Zhao, X. Oxidative Stress, Endocrine Disturbance, and Immune Interference in Humans Showed Relationships to Serum Bisphenol Concentrations in a Dense Industrial Area. Environ. Sci. Technol. 2021, 55, 1953–1963. [Google Scholar] [CrossRef]
- Singh, R.D.; Wager, J.L.; Scheidl, T.B.; Connors, L.T.; Easson, S.; Callaghan, M.A.; Alatorre-Hinojosa, S.; Swift, L.H.; Colarusso, P.; Jadli, A.; et al. Potentiation of Adipogenesis by Reactive Oxygen Species Is a Unifying Mechanism in the Proadipogenic Properties of Bisphenol A and Its New Structural Analogues. Antioxid. Redox Signal 2024, 40, 1–15. [Google Scholar] [CrossRef]
- Chen, J.; Ding, X.; Wu, R.; Tong, B.; Zhao, L.; Lv, H.; Meng, X.; Liu, Y.; Ren, B.; Li, J.; et al. Novel Sesquiterpene Glycoside from Loquat Leaf Alleviates Type 2 Diabetes Mellitus Combined with Nonalcoholic Fatty Liver Disease by Improving Insulin Resistance, Oxidative Stress, Inflammation, and Gut Microbiota Composition. J. Agric. Food Chem. 2021, 69, 14176–14191. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Jang, H.J.; Kim, S.; Choi, S.S.; Khim, K.W.; Eom, H.J.; Hyun, J.; Shin, K.J.; Chae, Y.C.; Kim, H.; et al. Hepatic MIR20B promotes nonalcoholic fatty liver disease by suppressing PPARA. Elife 2021, 10, e70472. [Google Scholar] [CrossRef]
- Qiu, W.; Yang, M.; Liu, J.; Xu, H.; Luo, S.; Wong, M.; Zheng, C. Bisphenol S-induced chronic inflammatory stress in liver via peroxisome proliferator-activated receptor gamma using fish in vivo and in vitro models. Environ. Pollut. 2019, 246, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Zhang, L.; Wu, T.; Fang, X.; Zhao, L. Echinacoside ameliorates alcohol-induced oxidative stress and hepatic steatosis by affecting SREBP1c/FASN pathway via PPARalpha. Food Chem. Toxicol. 2021, 148, 111956. [Google Scholar] [CrossRef] [PubMed]
- Maćczak, A.; Cyrkler, M.; Bukowska, B.; Michałowicz, J. Bisphenol A, bisphenol S, bisphenol F and bisphenol AF induce different oxidative stress and damage in human red blood cells (in vitro study). Toxicol. Vitr. 2017, 41, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, O.I.; Kurbatov, I.Y.; Arzumanian, V.A.; Ilgisonis, E.V.; Vakhrushev, I.V.; Lupatov, A.Y.; Ponomarenko, E.A.; Poverennaya, E.V. Exploring Dynamic Metabolome of the HepG2 Cell Line: Rise and Fall. Cells 2022, 11, 3548. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y. Impact of Extended and Combined Exposure of Bisphenol Compounds on Their Chromosome-Damaging Effect horizontal line Increased Potency and Shifted Mode of Action. Environ. Sci. Technol. 2023, 57, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Wu, Q.; Guo, L.; Ye, D.; Cao, Q.; Zhang, M.; Xian, Y.; Chen, M.; Yan, K.; Zheng, J. Pyridostigmine attenuated high-fat-diet induced liver injury by the reduction of mitochondrial damage and oxidative stress via α7nAChR and M3AChR. J. Biochem. Mol. Toxicol. 2024, 38, e23671. [Google Scholar] [CrossRef] [PubMed]
- Dhara, M.; Majumder, R.; Adhikari, L. Heliconia rostrata rhizomes mitigate chemical-induced liver injury by debilitating oxidative stress in HepG2 cells and rats. J. Ethnopharmacol. 2021, 275, 114113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, J.; Guo, Y.; Ye, H.; Yu, C.; Xu, C.; Xu, L.; Wu, S.; Sun, W.; Wei, H.; et al. Functional proteomic analysis of nonalcoholic fatty liver disease in rat models: Enoyl-coenzyme a hydratase down-regulation exacerbates hepatic steatosis. Hepatology 2010, 51, 1190–1199. [Google Scholar] [CrossRef]
- Huang, J.; Gao, T.; Zhang, H.; Wang, X. Association of obesity profiles and metabolic health status with liver injury among US adult population in NHANES 1999-2016. Sci. Rep. 2023, 13, 15958. [Google Scholar] [CrossRef]
- Jakubiak, G.K.; Osadnik, K.; Lejawa, M.; Kasperczyk, S.; Osadnik, T.; Pawlas, N. Oxidative Stress in Association with Metabolic Health and Obesity in Young Adults. Oxid. Med. Cell. Longev. 2021, 2021, 9987352. [Google Scholar] [CrossRef]
- Gianfrancesco, M.A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Lipid bilayer stress in obesity-linked inflammatory and metabolic disorders. Biochem. Pharmacol. 2018, 153, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Lugo, R.; Avila-Nava, A.; Pech-Aguilar, A.G.; Medina-Vera, I.; Guevara-Cruz, M.; Gutiérrez Solis, A.L. Relationship between lipid accumulation product and oxidative biomarkers by gender in adults from Yucatan, Mexico. Sci. Rep. 2022, 12, 14338. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, L.; Gai, Y.; Hong, Y.; Li, L.; Weng, L. Subchronic bisphenol S exposure affects liver function in mice involving oxidative damage. Regul. Toxicol. Pharmacol. 2018, 92, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Y.; Luo, L.; Kan, Y.Q.; Qin, M.L.; Li, H.T.; He, Q.Z.; Zeng, H.C. Puerarin Prevents Bisphenol S Induced Lipid Accumulation by Reducing Liver Lipid Synthesis and Promoting Lipid Metabolism in C57BL/6J Mice. Toxics 2023, 11, 736. [Google Scholar] [CrossRef]
- Balling, M.; Afzal, S.; Davey Smith, G.; Varbo, A.; Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. Elevated LDL Triglycerides and Atherosclerotic Risk. J. Am. Coll. Cardiol. 2023, 81, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef]
- Helies-Toussaint, C.; Peyre, L.; Costanzo, C.; Chagnon, M.C.; Rahmani, R. Is bisphenol S a safe substitute for bisphenol A in terms of metabolic function? An in vitro study. Toxicol. Appl. Pharmacol. 2014, 280, 224–235. [Google Scholar] [CrossRef]
- Mohan, S.; Jacob, J.; Malini, N.A.; Prabhakar, R.; Kayalakkakathu, R.G. Biochemical responses antioxidant defense mechanisms in Channa Striatus exposed to Bisphenol, S.J. Biochem. Mol. Toxicol. 2024, 38, e23651. [Google Scholar] [CrossRef] [PubMed]
- Sabry, R.; Nguyen, M.; Younes, S.; Favetta, L.A. BPA and its analogs increase oxidative stress levels in in vitro cultured granulosa cells by altering anti-oxidant enzymes expression. Mol. Cell Endocrinol. 2022, 545, 111574. [Google Scholar] [CrossRef]
- Song, R.; Hu, M.; Qin, X.; Qiu, L.; Wang, P.; Zhang, X.; Liu, R.; Wang, X. The Roles of Lipid Metabolism in the Pathogenesis of Chronic Diseases in the Elderly. Nutrients 2023, 15, 3433. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, J.; Tang, M.; Wang, X.; Fan, N.; Peng, Y. Fat mass and obesity-associated protein promotes liver steatosis by targeting PPARalpha. Lipids Health Dis. 2022, 21, 29. [Google Scholar] [CrossRef]
- Kumar, S.; Dhanwal, D.K. Central obesity & dyslipidemia in HIV patients on antiretroviral therapy. Indian J. Med. Res. 2018, 148, 366–368. [Google Scholar] [PubMed]
- Zhao, Q.; Yang, R.; Wang, J.; Hu, D.D.; Li, F. PPARalpha activation protects against cholestatic liver injury. Sci. Rep. 2017, 7, 9967. [Google Scholar]
- Lee, G.; Kim, Y.Y.; Jang, H.; Han, J.S.; Nahmgoong, H.; Park, Y.J.; Han, S.M.; Cho, C.; Lim, S.; Noh, J.R.; et al. SREBP1c-PARP1 axis tunes anti-senescence activity of adipocytes and ameliorates metabolic imbalance in obesity. Cell Metab. 2022, 34, 702–718. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, S.; Li, M.; Xiong, X.; Zhu, M.; Li, D.; Zhao, L.; Qian, L.; Zhai, L.; Li, J.; et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018, 9, 4770. [Google Scholar] [CrossRef]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Zeng, H.; Qin, H.; Liao, M.; Zheng, E.; Luo, X.; Xiao, A.; Li, Y.; Chen, L.; Wei, L.; Zhao, L.; et al. CD36 promotes de novo lipogenesis in hepatocytes through INSIG2-dependent SREBP1 processing. Mol. Metab. 2022, 57, 101428. [Google Scholar] [CrossRef] [PubMed]
- Nergiz-Unal, R.; Ulug, E.; Kisioglu, B.; Tamer, F.; Bodur, M.; Yalcimin, H.; Yuruk, A.A. Hepatic cholesterol synthesis and lipoprotein levels impaired by dietary fructose and saturated fatty acids in mice: Insight on PCSK9 and CD36. Nutrition 2020, 79–80, 110954. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Statin Residual Cardiovascular Disease Risk and Peroxisome Proliferator-Activated Receptor Alpha Agonists and Ezetimibe for Its Treatment. Int. J. Mol. Sci. 2022, 23, 3418. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yu, X.; Zhu, J.; Zeng, J.; Kuang, X.; Zhang, Y.; Tang, S.; Zhang, Q.; Yan, J.; Shen, H. Gastrodin ameliorates oxidative stress-induced RPE damage by facilitating autophagy and phagocytosis through PPARalpha-TFEB/CD36 signal pathway. Free Radic. Biol. Med. 2024, 224, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Kruppel-like factor 16 (KLF16) targets PPARalpha to improve steatohepatitis and insulin resistance. Gut 2021, 70, 2183–2195. [Google Scholar] [CrossRef]
- Sun, J.; Bian, Y.; Ma, Y.; Ali, W.; Wang, T.; Yuan, Y.; Gu, J.; Bian, J.; Liu, Z.; Zou, H. Melatonin alleviates cadmium-induced nonalcoholic fatty liver disease in ducks by alleviating autophagic flow arrest via PPAR-alpha and reducing oxidative stress. Poult. Sci. 2023, 102, 102835. [Google Scholar] [CrossRef] [PubMed]
- Hoepner, L.A. Bisphenol a: A narrative review of prenatal exposure effects on adipogenesis and childhood obesity via peroxisome proliferator-activated receptor gamma. Environ. Res. 2019, 173, 54–68. [Google Scholar] [CrossRef]
- Liang, X.; Liang, J.; Zhang, S.; Yan, H.; Luan, T. Di-2-ethylhexyl phthalate disrupts hepatic lipid metabolism in obese mice by activating the LXR/SREBP-1c and PPAR-alpha signaling pathways. Sci. Total Environ. 2024, 914, 1699. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequences (5′ → 3′) |
|---|---|---|
| FASN | Forward | CGGAGTCGCTTGAGTATA |
| Reverse | CACAGGGACCGAGTAATG | |
| PPARα | Forward | CAAGTGCCTGTCTGTCGG |
| Reverse | CAGGTAGGCTTCGTGGAT | |
| CD36 | Forward | ATTCTCATGCCAGTCGGA |
| Reverse | TTTGCTGCTGTTCTTTGC | |
| SREBP1C | Forward | ACAGTGACTTCCCTGGCCTAT |
| Reverse | GCATGGACGGGTACATCTTCAA | |
| CPT1B | Forward | AGACTGTGCGTTCCTGTA |
| Reverse | TTGGAGACGATGTAAAG | |
| GAPDH | Forward | CAGGAGGCATTGCTGATGAT |
| Reverse | GAAGGCTGGGGCTCATTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, K.-X.; Wu, Z.-Y.; Qin, M.-L.; Zeng, H.-C. Bisphenol S Induces Lipid Metabolism Disorders in HepG2 and SK-Hep-1 Cells via Oxidative Stress. Toxics 2025, 13, 44. https://doi.org/10.3390/toxics13010044
Lin K-X, Wu Z-Y, Qin M-L, Zeng H-C. Bisphenol S Induces Lipid Metabolism Disorders in HepG2 and SK-Hep-1 Cells via Oxidative Stress. Toxics. 2025; 13(1):44. https://doi.org/10.3390/toxics13010044
Chicago/Turabian StyleLin, Kai-Xing, Zi-Yao Wu, Mei-Lin Qin, and Huai-Cai Zeng. 2025. "Bisphenol S Induces Lipid Metabolism Disorders in HepG2 and SK-Hep-1 Cells via Oxidative Stress" Toxics 13, no. 1: 44. https://doi.org/10.3390/toxics13010044
APA StyleLin, K.-X., Wu, Z.-Y., Qin, M.-L., & Zeng, H.-C. (2025). Bisphenol S Induces Lipid Metabolism Disorders in HepG2 and SK-Hep-1 Cells via Oxidative Stress. Toxics, 13(1), 44. https://doi.org/10.3390/toxics13010044