
Effect of Volume Fraction of Carbon Nanotubes on Structure Formation in Polyacrylonitrile Nascent Fibers: Mesoscale Simulations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model and Simulation Method
2.1. Highlights of the DDFT
2.2. Model Description
2.3. Model Characteristic Scale
2.4. Interaction Parameters
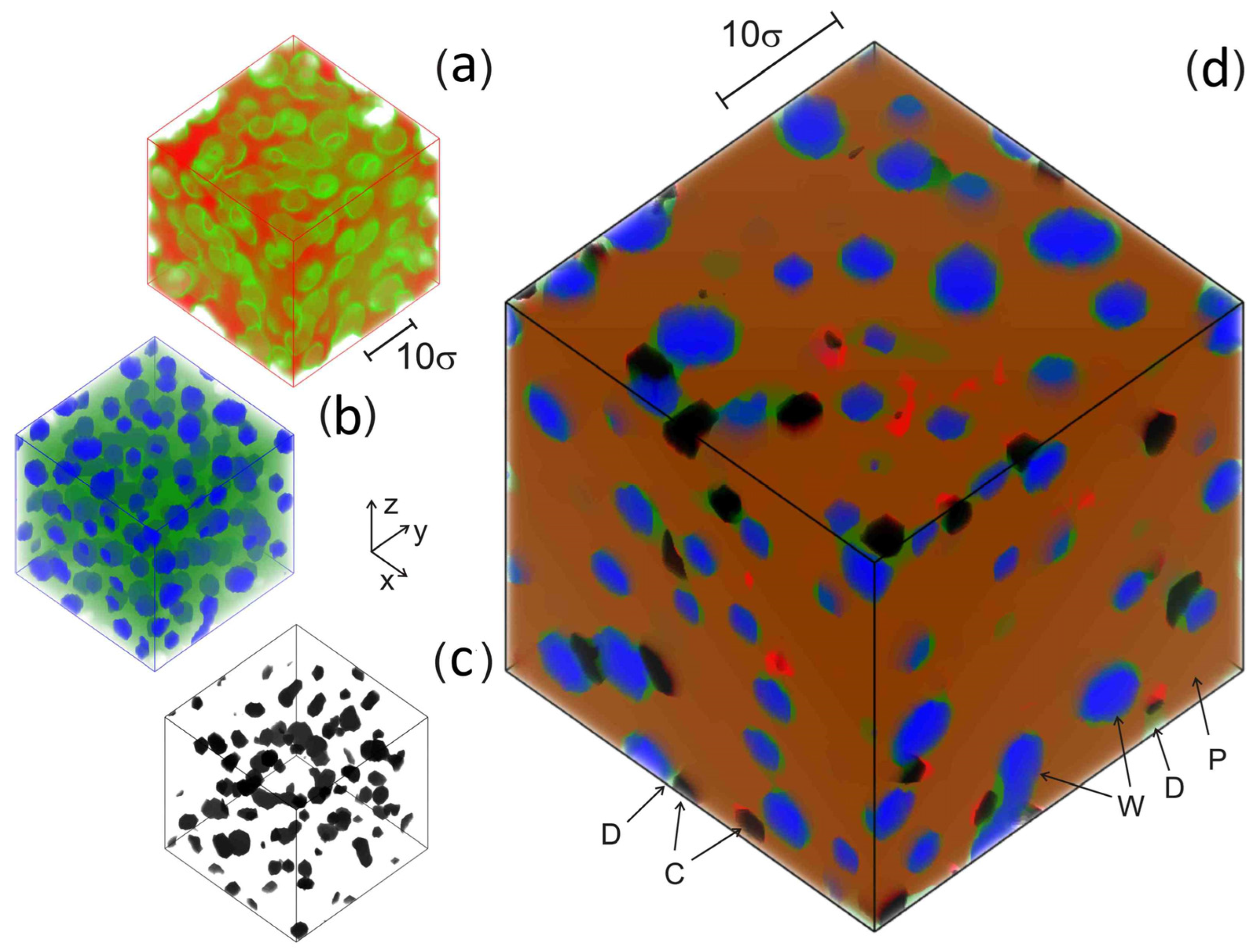
3. Calculation and System Characterization Methodology
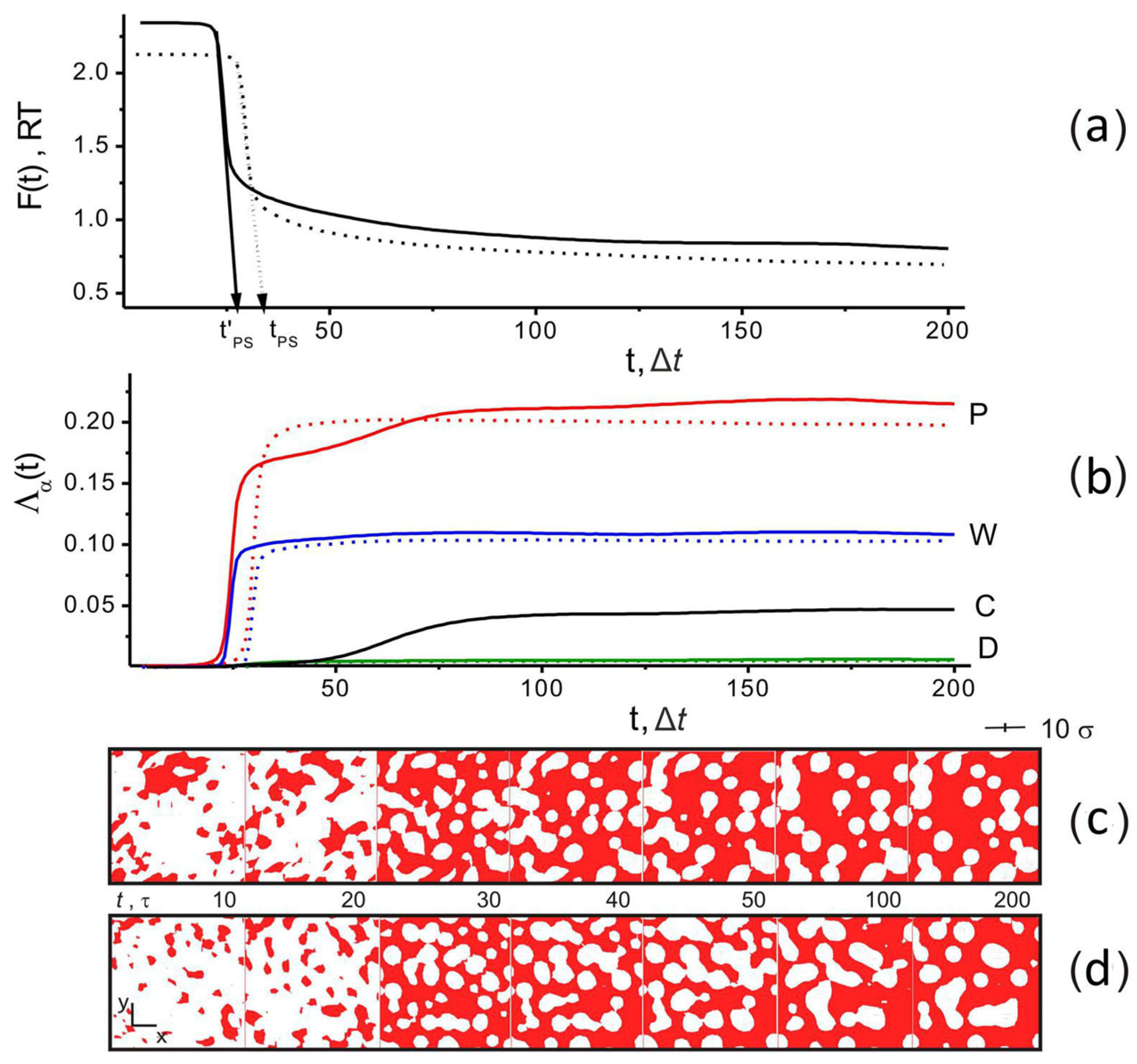
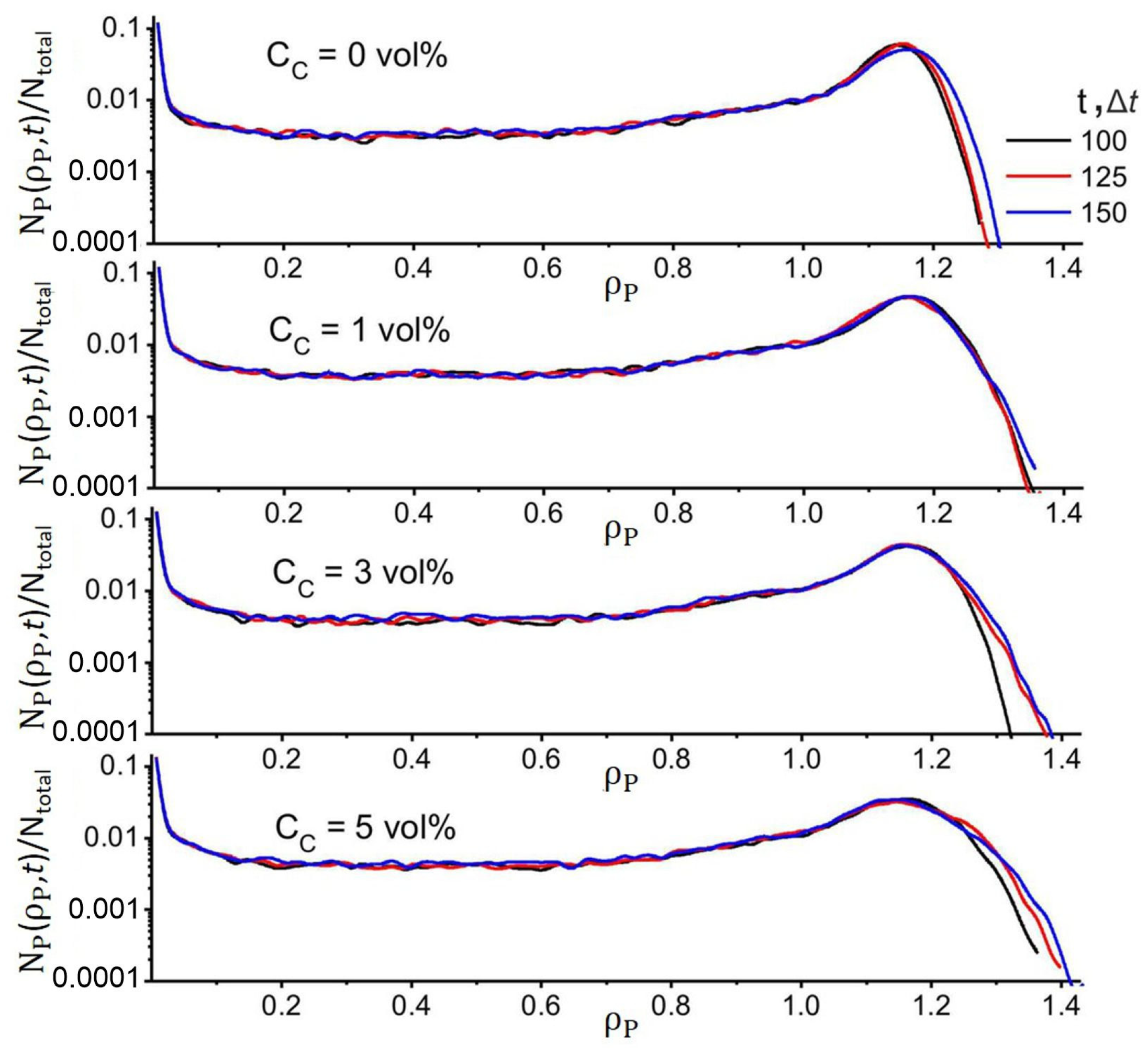
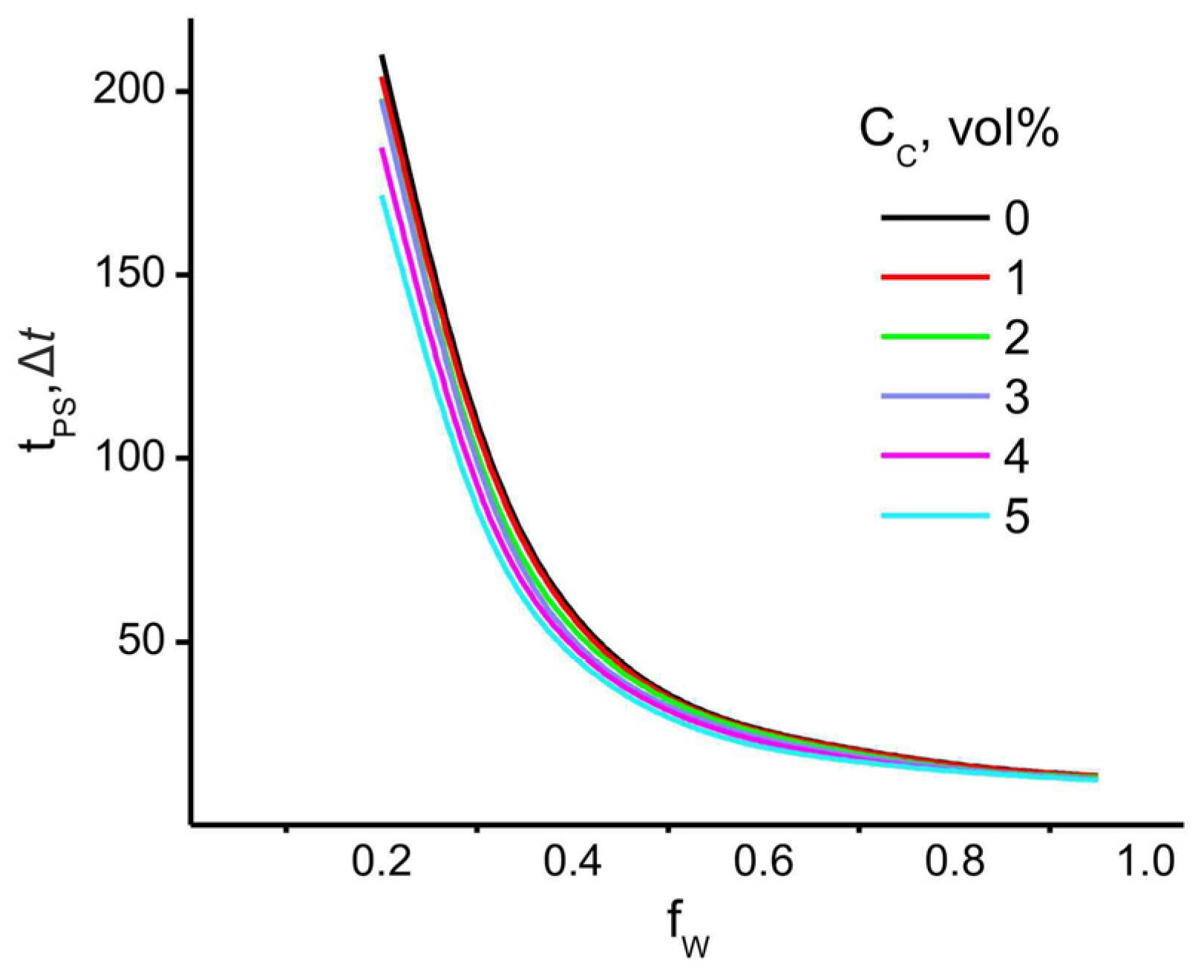
4. Results
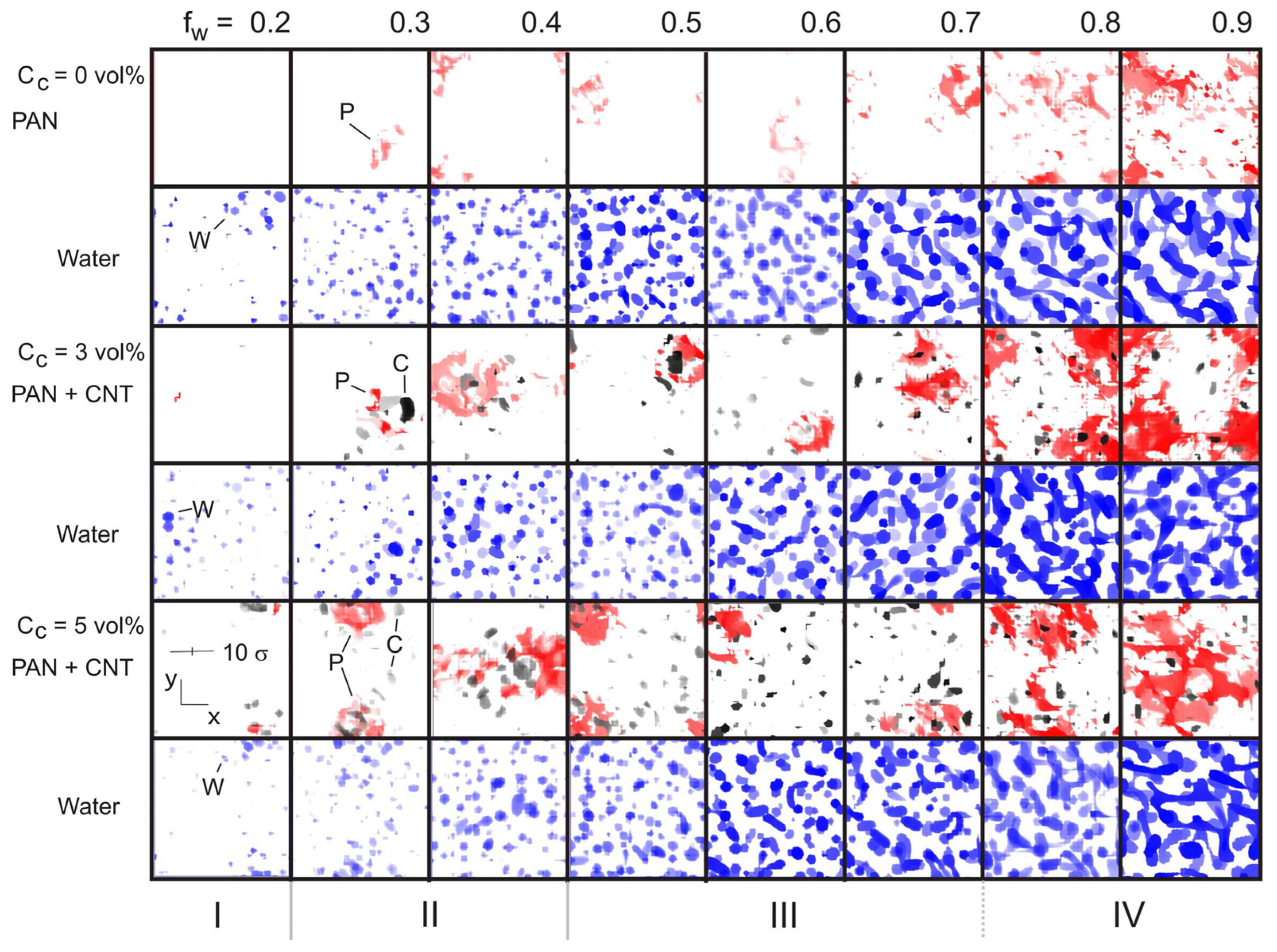
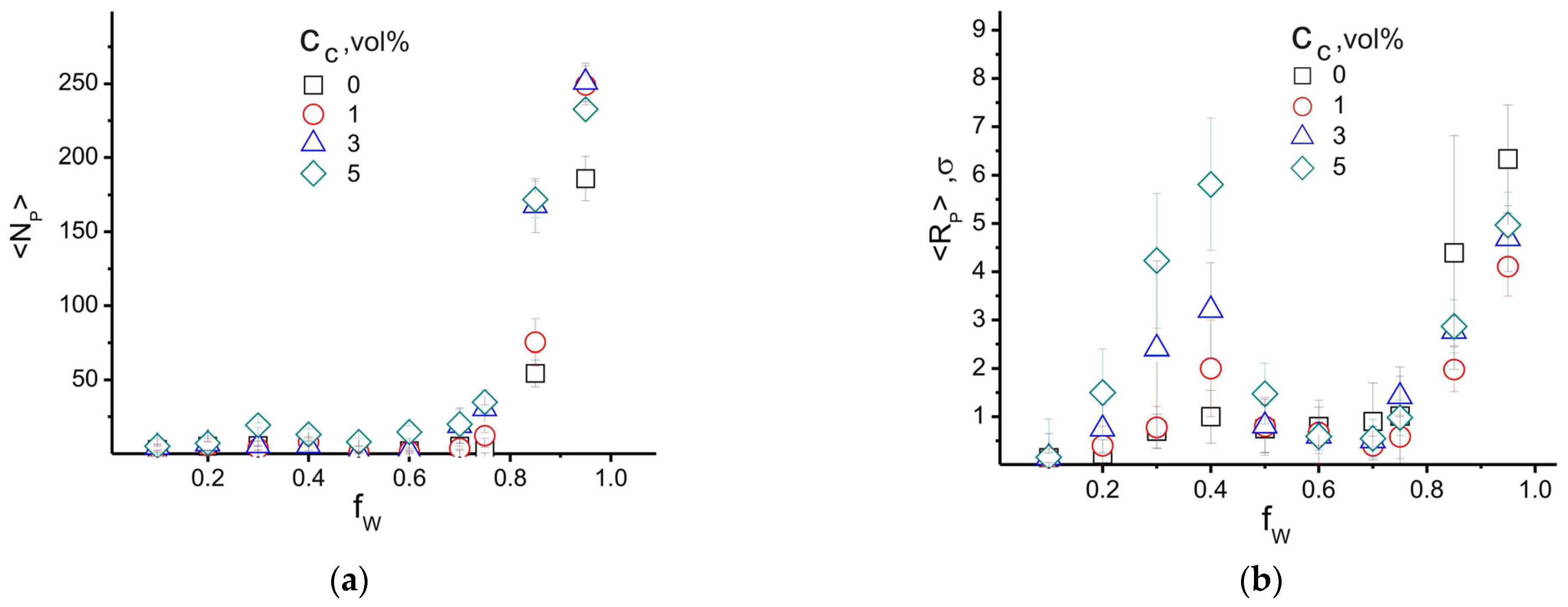
4.1. Effect of Filler and Mixed Solvent Quality on PAN Structure
4.2. Effect of Filler Surface Functionalization on the Structure of the Polymer Matrix
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chung, D.D.L. Carbon Fiber Composites; Butterworth-Heinemann: Newton, MA, USA, 1994. [Google Scholar]
- MInus, M.; Kumar, S. The Processing, Properties, and Structure of Carbon Fibers. JOM 2005, 57, 52–58. [Google Scholar] [CrossRef]
- Liu, Y.; Kumar, S. Recent Progress in Fabrication, Structure, and Properties of Carbon Fibers. Polym. Rev. 2012, 52, 234–258. [Google Scholar] [CrossRef]
- Ogale, A.A.; Zhang, M.; Jin, J. Recent Advances in Carbon Fibers Derived from Biobased Precursors. J. Appl. Polym. Sci. 2016, 133, 1–15. [Google Scholar] [CrossRef]
- Liu, H.C.; Chien, A.-T.; Newcomb, B.A.; Liu, Y.; Kumar, S. Processing, Structure, and Properties of Lignin- and CNT-Incorporated Polyacrylonitrile-Based Carbon Fibers. ACS Sustain. Chem. Eng. 2015, 3, 1943–1954. [Google Scholar] [CrossRef]
- Ahn, H.; Yeo, S.Y.; Lee, B.-S. Designing Materials and Processes for Strong Polyacrylonitrile Precursor Fibers. Polymers 2021, 13, 2863. [Google Scholar] [CrossRef]
- Frank, E.; Hermanutz, F.; Buchmeiser, M.R. Carbon Fibers: Precursors, Manufacturing, and Properties. Macromol. Mater. Eng. 2012, 297, 493–501. [Google Scholar] [CrossRef]
- Newcomb, B.A. Processing, Structure, and Properties of Carbon Fibers. Compos. Part A Appl. Sci. Manuf. 2016, 91, 262–282. [Google Scholar] [CrossRef]
- Nataraj, S.K.; Yang, K.S.; Aminabhavi, T.M. Polyacrylonitrile-Based nanofibers—A State-of-the-Art Review. Prog. Polym. Sci. 2012, 37, 487–513. [Google Scholar] [CrossRef]
- Gupta, A.K.; Paliwal, D.K.; Bajaj, P. Acrylic Precursors for Carbon Fibers. Polym. Rev. 1991, 31, 1–89. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A Review of Heat Treatment on Polyacrylonitrile Fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Hoffman, W.P.; Hurley, W.C.; Liu, P.M.; Owens, T.W. The Surface Topography of Non-Shear Treated Pitch and PAN Carbon Fibers as Viewed by the STM. J. Mater. Res. 1991, 6, 1685–1694. [Google Scholar] [CrossRef]
- Frank, E.; Steudle, L.M.; Ingildeev, D.; Spörl, J.M.; Buchmeiser, M.R. Carbon Fibers: Precursor Systems, Processing, Structure, and Properties. Angew. Chem. Int. Ed. Engl. 2014, 53, 5262–5298. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.R. Diffusion during the Coagulation Step of Wet-Spinning. J. Appl. Polym. Sci. 1968, 12, 383–402. [Google Scholar] [CrossRef]
- Takahashi, M.; Watanabe, M. Studies on Acrylic Fiber. Sen Gakkaishi 1959, 15, 951–959. [Google Scholar] [CrossRef]
- Craig, J.P.; Knudsen, J.P.; Holland, V.F. Characterization of Acrylic Fiber Structure. Text. Res. J. 1962, 32, 435–448. [Google Scholar] [CrossRef]
- Bell, J.P.; Dumbleton, J.H. Changes in the Structure of Wet-Spun Acrylic Fibers during Processing. Text. Res. J. 1971, 41, 196–203. [Google Scholar] [CrossRef]
- Wang, W.; Murthy, N.S.; Chae, H.G.; Kumar, S. Structural Changes during Deformation in Carbon Nanotube-Reinforced Polyacrylonitrile Fibers. Polymer 2008, 49, 2133–2145. [Google Scholar] [CrossRef]
- Sreekumar, T.V.; Liu, T.; Min, B.G.; Guo, H.; Kumar, S.; Hauge, R.H.; Smalley, R.E. Polyacrylonitrile Single-walled Carbon Nanotube Composite Fibers. Adv. Mater. 2004, 16, 58–61. [Google Scholar] [CrossRef]
- Koganemaru, A.; Bin, Y.; Agari, Y.; Matsuo, M. Composites of Polyacrylonitrile and Multiwalled Carbon Nanotubes Prepared by Gelation/crystallization from Solution. Adv. Funct. Mater. 2004, 14, 842–850. [Google Scholar] [CrossRef]
- Ye, H.; Lam, H.; Titchenal, N.; Gogotsi, Y.; Ko, F. Reinforcement and Rupture Behavior of Carbon Nanotubes–polymer Nanofibers. Appl. Phys. Lett. 2004, 85, 1775–1777. [Google Scholar] [CrossRef]
- Chae, H.G.; Sreekumar, T.V.; Uchida, T.; Kumar, S. A Comparison of Reinforcement Efficiency of Various Types of Carbon Nanotubes in Polyacrylonitrile Fiber. Polymer 2005, 46, 10925–10935. [Google Scholar] [CrossRef]
- Guo, H.; Sreekumar, T.V.; Liu, T.; Minus, M.; Kumar, S. Structure and Properties of Polyacrylonitrile/Single Wall Carbon Nanotube Composite Films. Polymer 2005, 46, 3001–3005. [Google Scholar] [CrossRef]
- Chae, H.G.; Minus, M.L.; Kumar, S. Oriented and Exfoliated Single Wall Carbon Nanotubes in Polyacrylonitrile. Polymer 2006, 47, 3494–3504. [Google Scholar] [CrossRef]
- Chae, H.G.; Kumar, S. Materials Science. Making Strong Fibers. Science 2008, 319, 908–909. [Google Scholar] [CrossRef]
- Chae, H.G.; Choi, Y.H.; Minus, M.L.; Kumar, S. Carbon Nanotube Reinforced Small Diameter Polyacrylonitrile Based Carbon Fiber. Compos. Sci. Technol. 2009, 69, 406–413. [Google Scholar] [CrossRef]
- Guo, H.; Minus, M.L.; Jagannathan, S.; Kumar, S. Polyacrylonitrile/Carbon Nanotube Composite Films. ACS Appl. Mater. Interfaces 2010, 2, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Minus, M.L.; Chae, H.G.; Kumar, S. Processing, Structure, and Properties of PAN/MWNT Composite Fibers. Macromol. Mater. Eng. 2010, 295, 742–749. [Google Scholar] [CrossRef]
- Min, B.G.; Sreekumar, T.V.; Uchida, T.; Kumar, S. Oxidative Stabilization of PAN/SWNT Composite Fiber. Carbon N. Y. 2005, 43, 599–604. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, Z.; Zhu, M.; Mo, X.; Chen, D. Electrospun Carbon Nanotube Composite Nanofibres with Uniaxially Aligned Arrays. Nanotechnology 2007, 18, 115611. [Google Scholar] [CrossRef]
- Karpushkin, E.A.; Berkovich, A.K.; Artemov, M.V.; Sergeev, V.G. Rheological Properties of Polyacrylonitrile Solutions Containing Highly Dispersed Carbon Nanotubes. Polym. Sci. Ser. A 2014, 56, 681–686. [Google Scholar] [CrossRef]
- Khalatur, P.G. Molecular Dynamics Simulations in Polymer Science. Polym. Sci. A Compr. Ref. 2012, 1, 417–460. [Google Scholar] [CrossRef]
- Oh, S.C.; Wang, Y.S.; Yeo, Y.-K. Modeling and Simulation of the Coagulation Process of Poly(acrylonitrile) Wet-Spinning. Ind. Eng. Chem. Res. 1996, 35, 4796–4800. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.-G.; Dong, X.-G.; Liu, H.-Z. Study on the Coagulation Mechanism of Wet-Spinning PAN Fibers. J. Polym. Res. 2006, 13, 515–519. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, G.; Zhang, Y.; Zhao, J.; Pan, D. Diffusion Mechanism of As-spun Polyacrylonitrile Fiber in a Dimethyl Sulfoxide-water Coagulation Bath. J. Macromol. Sci. Part A Pure Appl. Chem. 2006, 43, 1711–1720. [Google Scholar] [CrossRef]
- Chen, J.; Ge, H.; Liu, H.; Li, G.; Wang, C. The Coagulation Process of Nascent Fibers in PAN Wet-Spinning. J. Wuhan Univ. Technol. 2010, 25, 200–205. [Google Scholar] [CrossRef]
- Zhou, P.; Lu, C.; Shi, J.; Li, K.; He, F.; Zhang, S.; Li, Y. Effect of Bath Concentration on Coagulation Kinetics at the Early Stage during Wet Spinning of PAN Copolymer Nascent Fibers. J. Macromol. Sci. Phys. 2011, 50, 1215–1225. [Google Scholar] [CrossRef]
- Xu, N.; Ding, Y.; Schiesser, W.E.; Kothare, M.V. Mathematical Simulation of Wet Spinning Coagulation Process: Dynamic Modeling and Numerical Results. AIChE J. 2016, 62, 3432–3440. [Google Scholar] [CrossRef]
- Pires, J.M.; de Oliveira, O.V.; Freitas, L.C.G.; Filho, E.A.d.S.; Prado, A.R. Molecular Dynamic Simulations of Polyacrylonitrile in Ethanol and Water Solvents. Comput. Theor. Chem. 2012, 995, 75–78. [Google Scholar] [CrossRef]
- Wu, Q.-Y.; Chen, X.-N.; Wan, L.-S.; Xu, Z.-K. Interactions between Polyacrylonitrile and Solvents: Density Functional Theory Study and Two-Dimensional Infrared Correlation Analysis. J. Phys. Chem. B 2012, 116, 8321–8330. [Google Scholar] [CrossRef]
- Pramanik, C.; Jamil, T.; Gissinger, J.R.; Guittet, D.; Arias-Monje, P.J.; Kumar, S.; Heinz, H. Polyacrylonitrile Interactions with Carbon Nanotubes in Solution: Conformations and Binding as a Function of Solvent, Temperature, and Concentration. Adv. Funct. Mater. 2019, 29, 1905247. [Google Scholar] [CrossRef]
- Tallury, S.S.; Pasquinelli, M.A. Molecular Dynamics Simulations of Flexible Polymer Chains Wrapping Single-Walled Carbon Nanotubes. J. Phys. Chem. B 2010, 114, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.I.; Cho, A.E.; Kumar, S.; Jang, S.S.; Kim, Y.-H. Origin and Control of Polyacrylonitrile Alignments on Carbon Nanotubes and Graphene Nanoribbons. Adv. Funct. Mater. 2018, 28, 1706970. [Google Scholar] [CrossRef]
- Heo, S.J.; Kim, K.H.; Han, B.; Chae, H.G.; Lee, S.G. Defect Structure Evolution of Polyacrylonitrile and Single Wall Carbon Nanotube Nanocomposites: A Molecular Dynamics Simulation Approach. Sci. Rep. 2020, 10, 11816. [Google Scholar] [CrossRef]
- Komarov, P.; Malyshev, M.; Baburkin, P.; Guseva, D. Mesoscale Simulations of Structure Formation in Polyacrylonitrile Nascent Fibers Induced by Binary Solvent Mixture. Int. J. Mol. Sci. 2023, 24, 9312. [Google Scholar] [CrossRef]
- Fraaije, J.G.E.M.; van Vlimmeren, B.A.C.; Maurits, N.M.; Postma, M.; Evers, O.A.; Hoffmann, C.; Altevogt, P.; Goldbeck-Wood, G. The Dynamic Mean-Field Density Functional Method and Its Application to the Mesoscopic Dynamics of Quenched Block Copolymer Melts. J. Chem. Phys. 1997, 106, 4260–4269. [Google Scholar] [CrossRef]
- Komarov, P.V.; Veselov, I.N.; Khalatur, P.G. Nanoscale morphology in sulfonated poly(ether ether ketone)-based ionomeric membranes: Mesoscale simulations. Polym. Sci. Ser. A 2010, 52, 191–208. [Google Scholar] [CrossRef]
- Komarov, P.V.; Veselov, I.N.; Chu, P.P.; Khalatur, P.G.; Khokhlov, A.R. Atomistic and Mesoscale Simulation of Polymer Electrolyte Membranes Based on Sulfonated Poly(ether Ether Ketone). Chem. Phys. Lett. 2010, 487, 291–296. [Google Scholar] [CrossRef]
- Komarov, P.V.; Veselov, I.N.; Chu, P.P.; Khalatur, P.G. Mesoscale simulation of polymer electrolyte membranes based on sulfonated poly(ether ether ketone) and Nafion. Soft Matter. 2010, 6, 3939–3956. [Google Scholar] [CrossRef]
- Altevogt, P. The MesoDyn Project: Software for Mesoscale Chemical Engineering. Theochem 1999, 463, 139–143. [Google Scholar] [CrossRef]
- Wescott, J.T.; Qi, Y.; Subramanian, L.; Capehart, T.W. Mesoscale Simulation of Morphology in Hydrated Perfluorosulfonic Acid Membranes. J. Chem. Phys. 2006, 124, 134702. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1988. [Google Scholar]
- Huggins, M.L. Solutions of Long Chain Compounds. J. Chem. Phys. 1941, 9, 440. [Google Scholar] [CrossRef]
- Flory, P.J. Fifteenth Spiers Memorial Lecture. Thermodynamics of Polymer Solutions. Discuss. Faraday Soc. 1970, 49, 7–29. [Google Scholar] [CrossRef]
- Müller-Plathe, F. Scale-Hopping in Computer Simulations of Polymers. Soft Mater. 2002, 1, 1–31. [Google Scholar] [CrossRef]
- Tan, L.; Chen, H.; Pan, D.; Pan, N. Investigating the Spinnability in the Dry-Jet Wet Spinning of PAN Precursor Fiber. J. Appl. Polym. Sci. 2008, 110, 1997–2000. [Google Scholar] [CrossRef]
- Huang, X. Fabrication and Properties of Carbon Fibers. Materials 2009, 2, 2369–2403. [Google Scholar] [CrossRef]
- Tan, L.; Chen, H.; Pan, D.; Pan, N. Investigation into the Gelation and Crystallization of Polyacrylonitrile. Eur. Polym. J. 2009, 45, 1617–1624. [Google Scholar] [CrossRef]
- Chen, J.; Ge, H.-Y.; Dong, X.-G.; Wang, C.-G. The Formation of Polyacrylonitrile Nascent Fibers in Wet-Spinning Process. J. Appl. Polym. Sci. 2007, 106, 692–696. [Google Scholar] [CrossRef]
- Ge, H.; Liu, H.; Chen, J.; Wang, C. The Skin-Core Structure of Poly(acrylonitrile-Itaconic Acid) Precursor Fibers in Wet-Spinning. J. Appl. Polym. Sci. 2008, 108, 947–952. [Google Scholar] [CrossRef]
- Peng, G.-Q.; Zhang, X.-H.; Wen, Y.-F.; Yang, Y.-G.; Liu, L. Effect of Coagulation Bath DMSO Concentration on the Structure and Properties of Polyacrylonitrile (PAN) Nascent Fibers during Wet-Spinning. J. Macromol. Sci. Phys. 2008, 47, 1130–1141. [Google Scholar] [CrossRef]
- Bicerano, J. Prediction of Polymer Properties, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Kamide, K.; Miyazaki, Y.; Kobayashi, H. Light Scattering and Viscometric Study on Polyacrylonitrile in Dimethylformamide, Ethylene Carbonate/water Mixture, and Aqueous Nitric Acid. Polym. J. 1985, 17, 607–619. [Google Scholar] [CrossRef]
- Wu, S. Predicting Chain Conformation and Entanglement of Polymers from Chemical Structure. Polym. Eng. Sci. 1992, 32, 823–830. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, S.-J.; Park, S.; Kim, S.; Kern, N.R.; Lee, J.; Im, W. CHARMM-GUI Polymer Builder for Modeling and Simulation of Synthetic Polymers. J. Chem. Theory Comput. 2021, 17, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Abderaman, M.B.; Talla, K.; Gueye, E.-H.O.; Dione, A.N.; Faye, O.; Beye, A.C. A molecular dynamics simulation study on the miscibility of polyglycolide with polyacrylonitrile. J. Polym. Biopolym. Phys. Chem. 2018, 6, 31–38. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Miller-Chou, B.A.; Koenig, J.L. A Review of Polymer Dissolution. Prog. Polym. Sci. 2003, 28, 1223–1270. [Google Scholar] [CrossRef]
- Belmares, M.; Blanco, M.; Goddard, W.A., 3rd; Ross, R.B.; Caldwell, G.; Chou, S.-H.; Pham, J.; Olofson, P.M.; Thomas, C. Hildebrand and Hansen Solubility Parameters from Molecular Dynamics with Applications to Electronic Nose Polymer Sensors. J. Comput. Chem. 2004, 25, 1814–1826. [Google Scholar] [CrossRef]
- Askadskiĭ, A.A. Computational Materials Science of Polymers; Cambridge Int Science Publishing: Cambridge, UK, 2003. [Google Scholar]
- Hildebrand, J.H.; Scott, R.L. The Solubility of Nonelectrolytes, 3rd ed.; Reinhold Publishing Corporation: New York, NY, USA, 1950. [Google Scholar]
- Malkin, A.; Ilyin, S.; Roumyantseva, T.; Kulichikhin, V. Rheological Evidence of Gel Formation in Dilute Poly(acrylonitrile) Solutions. Macromolecules 2013, 46, 257–266. [Google Scholar] [CrossRef]
- Eom, Y.; Kim, B.C. Solubility Parameter-Based Analysis of Polyacrylonitrile Solutions in N,N-Dimethyl Formamide and Dimethyl Sulfoxide. Polymer 2014, 55, 2570–2577. [Google Scholar] [CrossRef]
- Barton, A.F.M. Solubility Parameters. Chem. Rev. 1975, 75, 731–753. [Google Scholar] [CrossRef]
- Alshehri, S.; Hussain, A.; Ahsan, M.N.; Ali, R.; Siddique, M.U.M. Thermodynamic, Computational Solubility Parameters in Organic Solvents and GastroPlus Based Prediction of Ketoconazole. ACS Omega 2021, 6, 5033–5045. [Google Scholar] [CrossRef]
- Wypych, G. Handbook of Solvents; ChemTec Publishing: Toronto, ON, Canada, 2001. [Google Scholar]
- Detriche, S.; Zorzini, G.; Colomer, J.F.; Fonseca, A.; Nagy, J.B. Application of the Hansen Solubility Parameters Theory to Carbon Nanotubes. J. Nanosci. Nanotechnol. 2008, 8, 6082–6092. [Google Scholar] [CrossRef]
- Bergin, S.D.; Sun, Z.; Rickard, D.; Streich, P.V.; Hamilton, J.P.; Coleman, J.N. Multicomponent Solubility Parameters for Single-Walled Carbon Nanotube-Solvent Mixtures. ACS Nano 2009, 3, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Çavuş, S.; Çakal, E.; Sevgili, L.M. Solvent Dependent Swelling Behaviour of poly(N-Vinylcaprolactam) and poly(N-Vinylcaprolactam-Co-Itaconic Acid) Gels and Determination of Solubility Parameters. Chem. Pap. 2015, 69, 1367–1377. [Google Scholar] [CrossRef]
- Tan, L.; Liu, S.; Pan, D. Water Effect on the Gelation Behavior of Polyacrylonitrile/dimethyl Sulfoxide Solution. Colloids Surf. A Physicochem. Eng. Asp. 2009, 340, 168–173. [Google Scholar] [CrossRef]
- Catalán, J.; Díaz, C.; García-Blanco, F. Characterization of Binary Solvent Mixtures of DMSO with Water and Other Cosolvents. J. Org. Chem. 2001, 66, 5846–5852. [Google Scholar] [CrossRef]
- Akhukov, M.A.; Chorkov, V.A.; Gavrilov, A.A.; Guseva, D.V.; Khalatur, P.G.; Khokhlov, A.R.; Kniznik, A.A.; Komarov, P.V.; Okun, M.V.; Potapkin, B.V.; et al. MULTICOMP Package for Multilevel Simulation of Polymer Nanocomposites. Comput. Mater. Sci. 2023, 216, 111832. [Google Scholar] [CrossRef]
- Sun, H. Ab Initio Calculations and Force Field Development for Computer Simulation of Polysilanes. Macromolecules 1995, 28, 701–712. [Google Scholar] [CrossRef]
- Morgan, P. Carbon Fibers and Their Composites, 1st ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar] [CrossRef]
- Hua, Z.; Zhong, Y.J.; Li, D.F. Pores in PAN-based carbon fiber and effects of pore defects on mechanical properties. Mater. Sci. Forum 2007, 546, 1665–1668. [Google Scholar] [CrossRef]
- Arbab, S.; Noorpanah, P.; Mohammadi, N.; Soleimani, M. Designing Index of Void Structure and Tensile Properties in Wet-spun Polyacrylonitrile (PAN) Fiber. I. Effect of Dope Polymer or Nonsolvent Concentration. J. Appl. Polym. Sci. 2008, 109, 3461–3469. [Google Scholar] [CrossRef]
- Han, C.D.; Segal, L. A Study of Fiber Extrusion in Wet Spinning. II. Effects of Spinning Conditions on Fiber Formation. J. Appl. Polym. Sci. 1970, 14, 2999–3019. [Google Scholar] [CrossRef]
- Yi, K.; Li, Q.F.; Zhang, L.; Li, N.; Zhou, Y.; Ryu, S.K.; Jin, R.G. Diffusion Coefficients of Dimethyl Sulphoxide (DMSO) and H2O in PAN Wet Spinning and Its Influence on Morphology of Nascent Polyacrylonitrile (PAN) Fiber. J. Eng. Fiber. Fabr. 2013, 8, 155892501300800. [Google Scholar] [CrossRef]
- Wang, B.; Li, C.; Cao, W. Effect of Polyacrylonitrile Precursor Orientation on the Structures and Properties of Thermally Stabilized Carbon Fiber. Materials 2021, 14, 3237. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Liu, J.; Wang, X.; Yan, K.; Zhang, A.; Yang, J.; Chen, L.; Liang, J. Structural Transformation of Polyacrylonitrile (PAN) Fibers during Rapid Thermal Pretreatment in Nitrogen Atmosphere. Polymers 2020, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Bashir, Z. The Hexagonal Mesophase in Atactic Polyacrylonitrile: A New Interpretation of the Phase Transitions in the Polymer. J. Macromol. Sci. Phys. 2001, 40, 41–67. [Google Scholar] [CrossRef]
- Knudsen, J.P. The Influence of Coagulation Variables on the Structure and Physical Properties of an Acrylic Fiber. Text. Res. J. 1963, 33, 13–20. [Google Scholar] [CrossRef]
- Qin, J. The Enhancement of Microvoids in Acrylic Fibers. J. Appl. Polym. Sci. 1992, 44, 1095–1105. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Wang, C.-G.; Yu, M.-J. Effects of Different Coagulation Conditions on Polyacrylonitrile Fibers Wet Spun in a System of Dimethylsulphoxide and Water. J. Appl. Polym. Sci. 2007, 104, 3723–3729. [Google Scholar] [CrossRef]
- Dong, R.; Keuser, M.; Zeng, X.; Zhao, J.; Zhang, Y.; Wu, C.; Pan, D. Viscometric Measurement of the Thermodynamics of PAN terpolymer/DMSO/water System and Effect of Fiber-Forming Conditions on the Morphology of PAN Precursor. J. Polym. Sci. B Polym. Phys. 2008, 46, 1997–2011. [Google Scholar] [CrossRef]
- Dong, R.; Zhao, J.; Zhang, Y.; Pan, D. Morphology Control of Polyacrylonitrile (PAN) Fibers by Phase Separation Technique. J. Polym. Sci. B Polym. Phys. 2009, 47, 261–275. [Google Scholar] [CrossRef]
- Arbab, S.; Zeinolebadi, A.; Noorpanah, P. Exploring the Thermodynamic Aspects of Structure Formation during Wet-Spinning of Polyacrylonitrile Fibres. In Methodologies and Applications for Chemoinformatics and Chemical Engineering; IGI Global: Hershey, PA, USA, 2013; p. 138. [Google Scholar] [CrossRef]
- Ji, B.H. Effect of coagulation bath concentration on the structure and properties of polyacrylonitrile as-spun fibers during wet-spinning. Adv. Mater. Res. 2011, 287, 1832–1836. [Google Scholar] [CrossRef]
- Morris, E.; Weisenberger, M.; Rice, G. Properties of PAN Fibers Solution Spun into a Chilled Coagulation Bath at High Solvent Compositions. Fibers 2015, 3, 560–574. [Google Scholar] [CrossRef]
- Samimi-Sohrforozani, E.; Azimi, S.; Abolhasani, A.; Malekian, S.; Arbab, S.; Zendehdel, M.; Abolhasani, M.M.; Yaghoobi Nia, N. Development of Porous Polyacrylonitrile Composite Fibers: New Precursor Fibers with High Thermal Stability. Electron. Mater. Lett. 2021, 2, 454–465. [Google Scholar] [CrossRef]
- Yushkin, A.; Basko, A.; Balynin, A.; Efimov, M.; Lebedeva, T.; Ilyasova, A.; Pochivalov, K.; Volkov, A. Effect of Acetone as Co-Solvent on Fabrication of Polyacrylonitrile Ultrafiltration Membranes by Non-Solvent Induced Phase Separation. Polymers 2022, 14, 4603. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kang, S.; Lee, H.; Kwak, D.-B.; Ou, Q.; Pei, C.; Pui, D.Y.H. Nanofiber Filter Performance Improvement: Nanofiber Layer Uniformity and Branched Nanofiber. Aerosol Air Qual. Res. 2020, 20, 80–88. [Google Scholar] [CrossRef]
- Cheng, X.; Zhao, L.; Zhang, Z.; Deng, C.; Li, C.; Du, Y.; Shi, J.; Zhu, M. Highly Efficient, Low-Resistant, Well-Ordered PAN Nanofiber Membranes for Air Filtration. Colloids Surf. A Physicochem. Eng. Asp. 2022, 655, 130302. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komarov, P.; Malyshev, M.; Baburkin, P.; Guseva, D. Effect of Volume Fraction of Carbon Nanotubes on Structure Formation in Polyacrylonitrile Nascent Fibers: Mesoscale Simulations. ChemEngineering 2024, 8, 97. https://doi.org/10.3390/chemengineering8050097
Komarov P, Malyshev M, Baburkin P, Guseva D. Effect of Volume Fraction of Carbon Nanotubes on Structure Formation in Polyacrylonitrile Nascent Fibers: Mesoscale Simulations. ChemEngineering. 2024; 8(5):97. https://doi.org/10.3390/chemengineering8050097
Chicago/Turabian StyleKomarov, Pavel, Maxim Malyshev, Pavel Baburkin, and Daria Guseva. 2024. "Effect of Volume Fraction of Carbon Nanotubes on Structure Formation in Polyacrylonitrile Nascent Fibers: Mesoscale Simulations" ChemEngineering 8, no. 5: 97. https://doi.org/10.3390/chemengineering8050097
APA StyleKomarov, P., Malyshev, M., Baburkin, P., & Guseva, D. (2024). Effect of Volume Fraction of Carbon Nanotubes on Structure Formation in Polyacrylonitrile Nascent Fibers: Mesoscale Simulations. ChemEngineering, 8(5), 97. https://doi.org/10.3390/chemengineering8050097