
Liver Ultrasound Histotripsy: Novel Analysis of the Histotripsy Site Cell Constituents with Implications for Histotripsy Application in Cell Transplantation and Cancer Therapy
 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Overall Experimental Method
2.1.1. HIFU Set Up
2.1.2. Modelling to Define the Histotripsy Parameters
2.1.3. Organ Perfusion, Histotripsy Lesion Formation and Assessment Perfusion Set Up
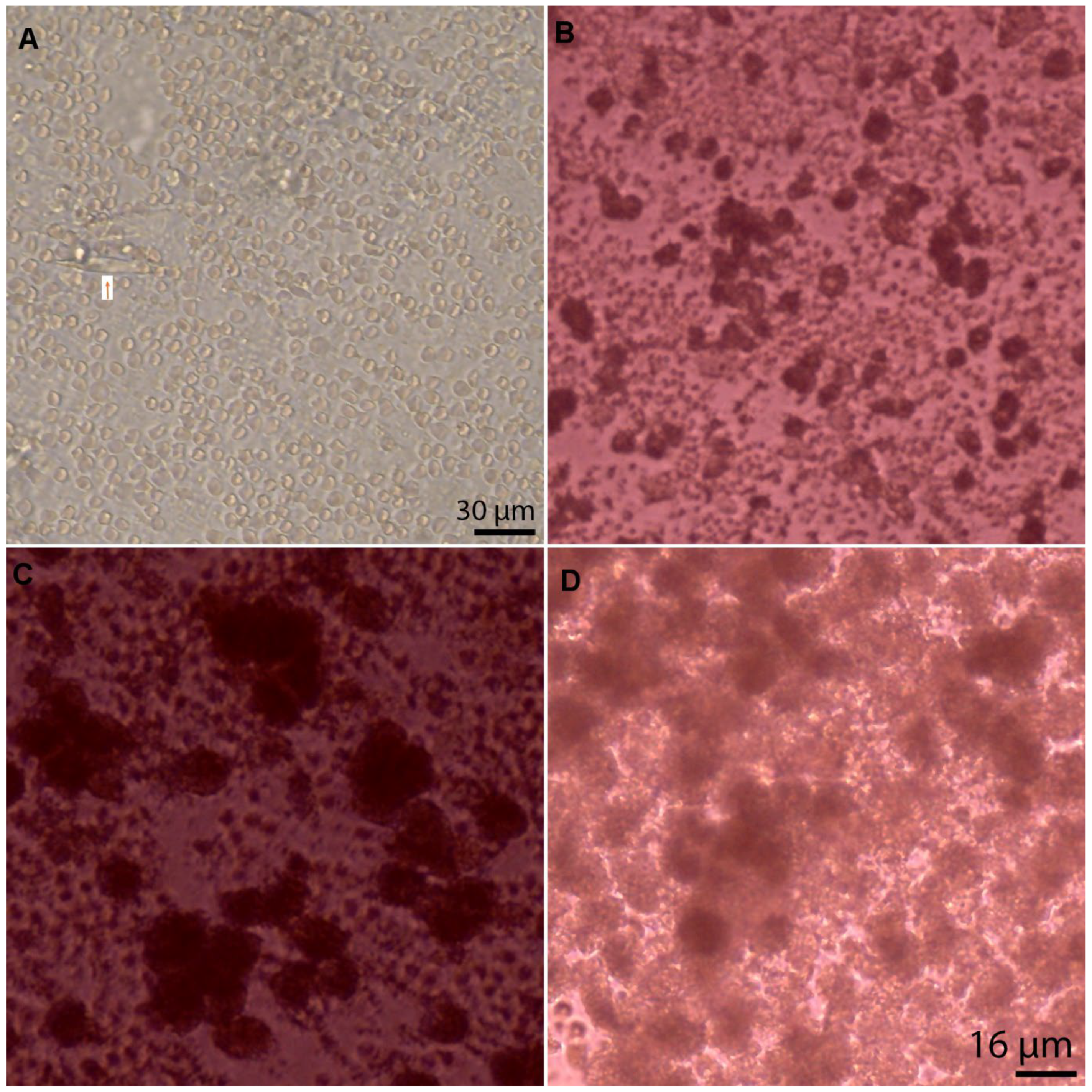
2.2. Pre-Culture Light Microscopy
2.3. Cell Culture: Cell Morphology and Growth
2.4. Phalloidin/DAPI Staining
2.5. Live-Dead Assay
2.6. Morphology Assessment
2.7. Cell Titre-Glo Metabolic Assay
2.8. Histological Evaluation of the Excised Histotripsy Sites
2.9. ImageJ Software
2.10. Statistical Methods
3. Results
3.1. Organ Perfusion and Viability, Histotripsy Lesion Creation and Core Aspiration
3.2. Histology of Lesions
3.3. Cell Culture
3.4. Cell Type, Appearances, and Numbers at Baseline
3.5. Cell Division and Viability in Culture
4. Discussion
4.1. Treatment Dose and the Nature of the Liver Lesion Based on Histology
4.2. The Use of the Viable Perfused Organ as a Model of In Vivo Histotripsy
4.3. The Cell Isolates, Culture, and Cell Viability
5. Limitation of Current Study, Further Evaluation, and Possible Implications for Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iansante, V.; Mitry, R.R.; Filippi, C.; Fitzpatrick, E.; Dhawan, A. Human hepatocyte transplantation for liver disease: Current status and future perspectives. Pediatr. Res. 2018, 83, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Puppi, J.; Hughes, R.D.; Mitry, R.R. Human hepatocyte transplantation: Current experience and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 288–298. [Google Scholar] [CrossRef]
- Hughes, R.D.; Mitry, R.R.; Dhawan, A.; Lehec, S.C.; Girlanda, R.; Rela, M.; Heaton, N.D.; Muiesan, P. Isolation of hepatocytes from livers from non-heart-beating donors for cell transplantation. Liver Transplant. 2006, 12, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.J.; Soriano, H.E. Liver cell transplantation: The road to clinical application. J. Lab. Clin. Med. 2001, 138, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Mitry, R.R.; Hughes, R.D. Hepatocyte transplantation for liver-based metabolic disorders. J. Inherit. Metab. Dis. 2006, 29, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Strom, S.C. Human Hepatocyte Transplantation: Worldwide Results. Transplantation 2006, 82, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Puppi, J.; Strom, S.C.; Hughes, R.D.; Bansal, S.; Castell, J.V.; Dagher, I.; Ellis, E.C.S.; Nowak, G.; Ericzon, B.-G.; Fox, I.J.; et al. Improving the Techniques for Human Hepatocyte Transplantation: Report from a Consensus Meeting in London. Cell Transplant. 2012, 21, 1–10. [Google Scholar] [CrossRef]
- Dhawan, A. Clinical human hepatocyte transplantation: Current status and challenges. Liver Transplant. 2015, 21, S39–S44. [Google Scholar] [CrossRef]
- Mitry, R.R.; Hughes, R.D.; Dhawan, A. Progress in human hepatocytes: Isolation, culture & cryopreservation. Semin. Cell Dev. Biol. 2002, 13, 463–467. [Google Scholar] [CrossRef]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells. J. Vis. Exp. 2016, 109, e53069. [Google Scholar] [CrossRef]
- Yu, Y.; Fisher, J.E.; Lillegard, J.B.; Rodysill, B.; Amiot, B.; Nyberg, S.L. Cell therapies for liver diseases. Liver Transplant. 2012, 18, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Khokhlova, T.D.; Buravkov, S.; Chernikov, V.; Kreider, W.; Partanen, A.; Farr, N.; Maxwell, A.; Schade, G.R.; Khokhlova, V.A. Mechanical decellularization of tissue volumes using boiling histotripsy. Phys. Med. Biol. 2018, 63, 235023. [Google Scholar] [CrossRef] [PubMed]
- Khokhlova, V.A.; Fowlkes, J.B.; Roberts, W.W.; Schade, G.R.; Xu, Z.; Khokhlova, T.D.; Hall, T.L.; Maxwell, A.D.; Wang, Y.-N.; Cain, C.A. Histotripsy methods in mechanical disintegration of tissue: Towards clinical applications. Int. J. Hyperth. 2015, 31, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Eranki, A.; Farr, N.; Partanen, A.; Sharma, K.V.; Chen, H.; Rossi, C.T.; Kothapalli, S.V.V.N.; Oetgen, M.; Kim, A.; Negussie, A.H.; et al. Boiling histotripsy lesion characterization on a clinical magnetic resonance imaging-guided high intensity focused ultrasound system. PLoS ONE 2017, 12, e0173867. [Google Scholar] [CrossRef]
- de Andrade, M.O.; Haqshenas, S.R.; Pahk, K.J.; Saffari, N. The effects of ultrasound pressure and temperature fields in millisecond bubble nucleation. Ultrason. Sonochem. 2019, 55, 262–272. [Google Scholar] [CrossRef]
- Pahk, K.J.; de Andrade, M.O.; Gélat, P.; Kim, H.; Saffari, N. Mechanical damage induced by the appearance of rectified bubble growth in a viscoelastic medium during boiling histotripsy exposure. Ultrason. Sonochem. 2019, 53, 164–177. [Google Scholar] [CrossRef]
- Pahk, K.J.; Gélat, P.; Sinden, D.; Dhar, D.K.; Saffari, N. Numerical and Experimental Study of Mechanisms Involved in Boiling Histotripsy. Ultrasound Med. Biol. 2017, 43, 2848–2861. [Google Scholar] [CrossRef]
- Pahk, K.J.; Gélat, P.; Kim, H.; Saffari, N. Bubble dynamics in boiling histotripsy. Ultrasound Med. Biol. 2018, 44, 2673–2696. [Google Scholar] [CrossRef]
- Pahk, K.J.; Mohammad, G.H.; Malago, M.; Saffari, N.; Dhar, D.K. A Novel Approach to Ultrasound-Mediated Tissue Decellularization and Intra-Hepatic Cell Delivery in Rats. Ultrasound Med. Biol. 2016, 42, 1958–1967. [Google Scholar] [CrossRef]
- Berry, M.N. High-yield preparation of isolated rat liver parenchymal cells: A biochemical and fine structural study. J. Cell Biol. 1969, 43, 506–520. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Liu, S.; Chen, Z.-G.; Zou, J.-Z.; Wu, F. Cavitation enhances coagulated size during pulsed high-intensity focussed ultrasound ablation in an isolated liver perfusion system. Int. J. Hyperth. 2017, 33, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Soneson, J.E. A User-Friendly Software Package for HIFU Simulation. AIP Conf. Proc. 2009, 1113, 165–169. [Google Scholar] [CrossRef]
- Khokhlova, T.D.; Canney, M.S.; Khokhlova, V.A.; Sapozhnikov, O.A.; Crum, L.A.; Bailey, M.R. Controlled tissue emulsification produced by high intensity focused ultrasound shock waves and millisecond boiling. J. Acoust. Soc. Am. 2011, 130, 3498–3510. [Google Scholar] [CrossRef]
- Canney, M.S.; Bailey, M.R.; Crum, L.A.; Khokhlova, V.A.; Sapozhnikov, O.A. Acoustic characterization of high intensity focused ultrasound fields: A combined measurement and modeling approach. J. Acoust. Soc. Am. 2008, 124, 2406–2420. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Mestre, T.; Carneiro, P.; Sahumbaiev, I.; Seruca, R.; Sanches, J.M. Blue intensity matters for cell cycle profiling in fluorescence DAPI-stained images. Lab. Investig. 2017, 97, 615–625. [Google Scholar] [CrossRef]
- Menyhárt, O.; Harami-Papp, H.; Sukumar, S.; Schäfer, R.; Magnani, L.; de Barrios, O.; Győrffy, B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2016, 1866, 300–319. [Google Scholar] [CrossRef]
- Sehmbi, A.S.; Froghi, S.; Oliveira De Andrade, M.; Saffari, N.; Fuller, B.; Quaglia, A.; Davidson, B. Systematic review of the role of high intensity focused ultrasound (HIFU) in treating malignant lesions of the hepatobiliary system. HPB 2021, 23, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Worlikar, T.; Vlaisavljevich, E.; Gerhardson, T.; Greve, J.; Wan, S.; Kuruvilla, S.; Lundt, J.; Ives, K.; Hall, T.; Welling, T.H.; et al. Histotripsy for Non-Invasive Ablation of Hepatocellular Carcinoma (HCC) Tumor in a Subcutaneous Xenograft Murine Model. In Proceedings of the 40th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Honolulu, HI, USA, 18–21 July 2018; pp. 6064–6067. [Google Scholar] [CrossRef]
- Ruger, L.N.; Hay, A.N.; Gannon, J.M.; Sheppard, H.O.; Coutermarsh-Ott, S.L.; Daniel, G.B.; Kierski, K.R.; Ciepluch, B.J.; Vlaisavljevich, E.; Tuohy, J.L. Histotripsy Ablation of Spontaneously Occurring Canine Bone Tumors. IEEE Trans. Biomed. Eng. 2023, 70, 331–342. [Google Scholar] [CrossRef]
- Vachaparambil, K.J.; Einarsrud, K.E. Explanation of Bubble Nucleation Mechanisms: A Gradient Theory Approach. J. Electrochem. Soc. 2018, 165, E504–E512. [Google Scholar] [CrossRef]
- Vlaisavljevich, E.; Owens, G.; Lundt, J.; Teofilovic, D.; Ives, K.; Duryea, A.; Bertolina, J.; Welling, T.H.; Xu, Z. Non-Invasive Liver Ablation Using Histotripsy: Preclinical Safety Study in an In Vivo Porcine Model. Ultrasound Med. Biol. 2017, 43, 1237–1251. [Google Scholar] [CrossRef]
- Khokhlova, T.D.; Schade, G.R.; Wang, Y.-N.; Buravkov, S.V.; Chernikov, V.P.; Simon, J.C.; Starr, F.; Maxwell, A.D.; Bailey, M.R.; Kreider, W.; et al. Pilot in vivo studies on transcutaneous boiling histotripsy in porcine liver and kidney. Sci. Rep. 2019, 9, 20176. [Google Scholar] [CrossRef] [PubMed]
- Panconesi, R.; Flores Carvalho, M.; Mueller, M.; Meierhofer, D.; Dutkowski, P.; Muiesan, P.; Schlegel, A. Viability Assessment in Liver Transplantation—What Is the Impact of Dynamic Organ Preservation? Biomedicines 2021, 9, 161. [Google Scholar] [CrossRef]
- Vlaisavljevich, E.; Greve, J.; Cheng, X.; Ives, K.; Shi, J.; Jin, L.; Arvidson, A.; Hall, T.; Welling, T.H.; Owens, G.; et al. Non-Invasive Ultrasound Liver Ablation Using Histotripsy: Chronic Study in an In Vivo Rodent Model. Ultrasound Med. Biol. 2016, 42, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Vlaisavljevich, E.; Cain, C.A.; Xu, Z. The effect of histotripsy on tissues with different mechanical properties. In Proceedings of the 2011 IEEE International Ultrasonics Symposium, Orlando, FL, USA, 18–21 October 2011; pp. 1490–1493. [Google Scholar]
- Vlaisavljevich, E.; Kim, Y.; Owens, G.; Roberts, W.; Cain, C.; Xu, Z. Effects of tissue mechanical properties on susceptibility to histotripsy-induced tissue damage. Phys. Med. Biol. 2014, 59, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Schurink, I.J.; Willemse, J.; Verstegen, M.M.A.; Laan, L.J.W.; Jonge, J. Long-Term Perfusion of the Liver Outside the Body: Warming Up for Ex Vivo Therapies? Hepatology 2020, 72, 1485–1487. [Google Scholar] [CrossRef]
- Hafez, T.; Fuller, B.J. Ch9 Organ Preservation for Transplantation. In Advances in Biopreservation; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar] [CrossRef]
- Drochmans, P. Isolation and subfractionation on ficoll gradients of adult rat hepatocytes. Size, morphology, and biochemical characteristics of cell fractions. J. Cell Biol. 1975, 66, 1–22. [Google Scholar] [CrossRef]
- Lee, S.M.L.; Schelcher, C.; Demmel, M.; Hauner, M.; Thasler, W.E. Isolation of Human Hepatocytes by a Two-Step Collagenase Perfusion Procedure. J. Vis. Exp. 2013, 79, e50615. [Google Scholar] [CrossRef]
- Hameed, A.M.; Laurence, J.M.; Lam, V.W.T.; Pleass, H.C.; Hawthorne, W.J. A systematic review and meta-analysis of cold in situ perfusion and preservation of the hepatic allograft: Working toward a unified approach. Liver Transplant. 2017, 23, 1615–1627. [Google Scholar] [CrossRef]
- Cabral, F.; Miller, C.M.; Kudrna, K.M.; Hass, B.E.; Daubendiek, J.G.; Kellar, B.M.; Harris, E.N. Purification of Hepatocytes and Sinusoidal Endothelial Cells from Mouse Liver Perfusion. J. Vis. Exp. 2018, 132, e56993. [Google Scholar] [CrossRef]
- Bhandari, R.N.B.; Riccalton, L.A.; Lewis, A.L.; Fry, J.R.; Hammond, A.H.; Tendler, S.J.B.; Shakesheff, K.M. Liver Tissue Engineering: A Role for Co-culture Systems in Modifying Hepatocyte Function and Viability. Tissue Eng. 2001, 7, 345–357. [Google Scholar] [CrossRef]
- Shulman, M.; Nahmias, Y. Long-Term Culture and Coculture of Primary Rat and Human Hepatocytes; Humana Press: Totowa, NJ, USA, 2012; pp. 287–302. [Google Scholar]
- Wang, G.; Zheng, Y.; Wang, Y.; Cai, Z.; Liao, N.; Liu, J.; Zhang, W. Co-culture system of hepatocytes and endothelial cells: Two in vitro approaches for enhancing liver-specific functions of hepatocytes. Cytotechnology 2018, 70, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Seo, W. Hepatic non-parenchymal cells: Master regulators of alcoholic liver disease? World J. Gastroenterol. 2016, 22, 1348. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Saghatolislam, F.; Koenig, S.; Unthan-Fechner, K.; Probst, I. Maintaining Hepatocyte Differentiation in vitro through Co-Culture with Hepatic Stellate Cells. In Vitro Cell. Dev. Biol. Anim. 2009, 45, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Balis, U.J.; Yarmush, M.L.; Toner, M. Effect of cell–cell interactions in preservation of cellular phenotype: Cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 1999, 13, 1883–1900. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.J.; Bhandari, R.; Barrett, D.A.; Bennett, A.J.; Fry, J.R.; Powe, D.; Thomson, B.J.; Shakesheff, K.M. The Effect of Three-Dimensional Co-Culture of Hepatocytes and Hepatic Stellate Cells on Key Hepatocyte Functions in vitro. Cells Tissues Organs 2005, 181, 67–79. [Google Scholar] [CrossRef]
- Kolios, G. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413. [Google Scholar] [CrossRef]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 574276. [Google Scholar] [CrossRef]
- Abbas, N.; Getachew, A.; You, K.; Shah, Z.; Chen, Y.; Tao, J.; Hussain, M.; Yang, F.; Zhuang, Y.; Xu, Y.; et al. Kupffer cells mediate the recruitment of hepatic stellate cells into the localized liver damage. Biochem. Biophys. Res. Commun. 2020, 529, 474–479. [Google Scholar] [CrossRef]
- Cheng, Q.-N.; Yang, X.; Wu, J.-F.; Ai, W.-B.; Ni, Y.-R. Interaction of non-parenchymal hepatocytes in the process of hepatic fibrosis (Review). Mol. Med. Rep. 2021, 23, 364. [Google Scholar] [CrossRef]
- Baze, A.; Parmentier, C.; Hendriks, D.F.; Hurrell, T.; Heyd, B.; Bachellier, P.; Schuster, C.; Ingelman-Sundberg, M.; Richert, L. Three-Dimensional Spheroid Primary Human Hepatocytes in Monoculture and Coculture with Nonparenchymal Cells. Tissue Eng. Part C Methods 2018, 24, 534–545. [Google Scholar] [CrossRef]
- Rashidi, H.; Luu, N.-T.; Alwahsh, S.M.; Ginai, M.; Alhaque, S.; Dong, H.; Tomaz, R.A.; Vernay, B.; Vigneswara, V.; Hallett, J.M.; et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch. Toxicol. 2018, 92, 3117–3129. [Google Scholar] [CrossRef] [PubMed]
- Goulet, F.; Normand, C.; Morin, O. Cellular interactions promote tissue-specific function, biomatrix deposition and junctional communication of primary cultured hepatocytes. Hepatology 1988, 8, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Crespo Yanguas, S.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Da Silva, T.C.; Veloso Alves Pereira, I.; Jaeschke, H.; Cogliati, B.; et al. Structure, Regulation and Function of Gap Junctions in Liver. Cell Commun. Adhes. 2015, 22, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Papeleu, P.; Snykers, S.; De Rop, E.; Henkens, T.; Chipman, J.K.; Rogiers, V.; Vanhaecke, T. Involvement of Cell Junctions in Hepatocyte Culture Functionality. Crit. Rev. Toxicol. 2006, 36, 299–318. [Google Scholar] [CrossRef]
- Ntonas, A.; Katsourakis, A.; Galanis, N.; Filo, E.; Noussios, G. Comparative Anatomical Study between the Human and Swine Liver and Its Importance in Xenotransplantation. Cureus 2020, 12, e9411. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Live | Dead | Total | % Live | |
|---|---|---|---|---|
| Day 1 | 1206 | 8690 | 9896 | 12.2 |
| Day 7 | 2022 | 2460 | 4482 | 45.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Froghi, S.; de Andrade, M.O.; Hadi, L.M.; Gelat, P.; Rashidi, H.; Quaglia, A.; Fuller, B.; Saffari, N.; Davidson, B. Liver Ultrasound Histotripsy: Novel Analysis of the Histotripsy Site Cell Constituents with Implications for Histotripsy Application in Cell Transplantation and Cancer Therapy. Bioengineering 2023, 10, 276. https://doi.org/10.3390/bioengineering10020276
Froghi S, de Andrade MO, Hadi LM, Gelat P, Rashidi H, Quaglia A, Fuller B, Saffari N, Davidson B. Liver Ultrasound Histotripsy: Novel Analysis of the Histotripsy Site Cell Constituents with Implications for Histotripsy Application in Cell Transplantation and Cancer Therapy. Bioengineering. 2023; 10(2):276. https://doi.org/10.3390/bioengineering10020276
Chicago/Turabian StyleFroghi, Saied, Matheus Oliveira de Andrade, Layla Mohammad Hadi, Pierre Gelat, Hassan Rashidi, Alberto Quaglia, Barry Fuller, Nader Saffari, and Brian Davidson. 2023. "Liver Ultrasound Histotripsy: Novel Analysis of the Histotripsy Site Cell Constituents with Implications for Histotripsy Application in Cell Transplantation and Cancer Therapy" Bioengineering 10, no. 2: 276. https://doi.org/10.3390/bioengineering10020276
APA StyleFroghi, S., de Andrade, M. O., Hadi, L. M., Gelat, P., Rashidi, H., Quaglia, A., Fuller, B., Saffari, N., & Davidson, B. (2023). Liver Ultrasound Histotripsy: Novel Analysis of the Histotripsy Site Cell Constituents with Implications for Histotripsy Application in Cell Transplantation and Cancer Therapy. Bioengineering, 10(2), 276. https://doi.org/10.3390/bioengineering10020276