
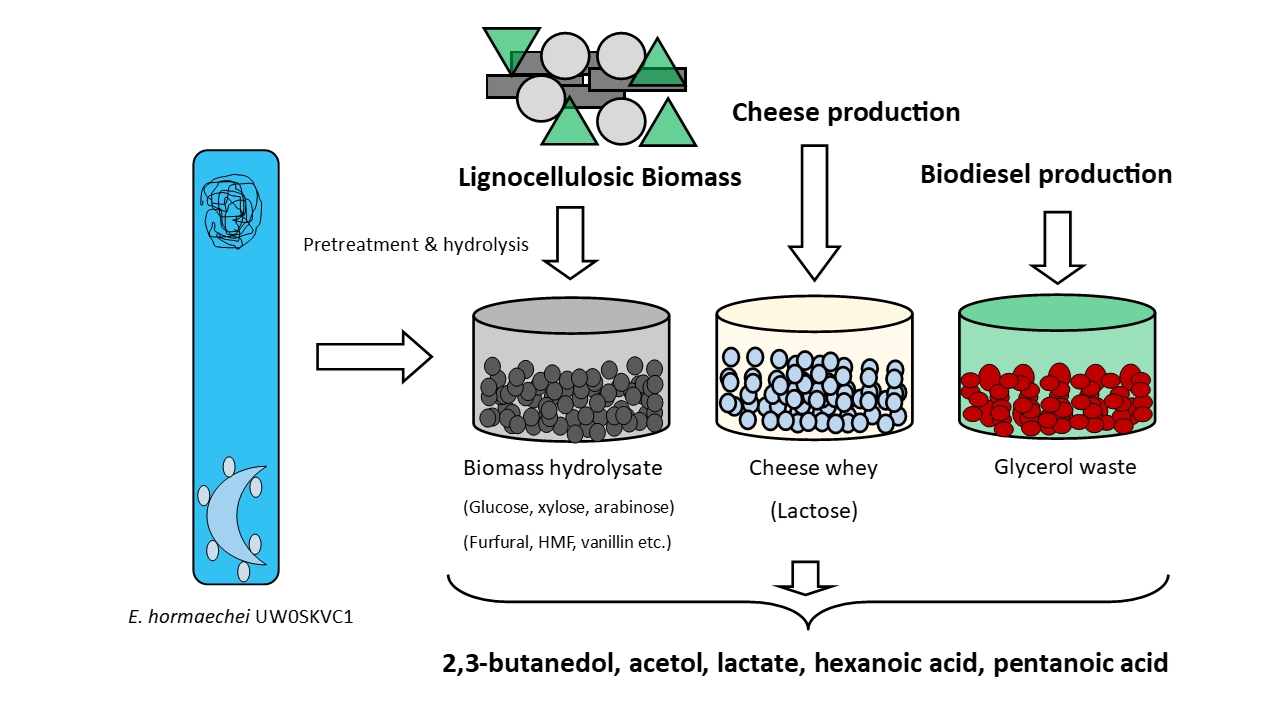
Whole-Genome Sequence and Fermentation Characteristics of Enterobacter hormaechei UW0SKVC1: A Promising Candidate for Detoxification of Lignocellulosic Biomass Hydrolysates and Production of Value-Added Chemicals



Abstract

1. Introduction
2. Material and Methods
2.1. Media and Culture Conditions
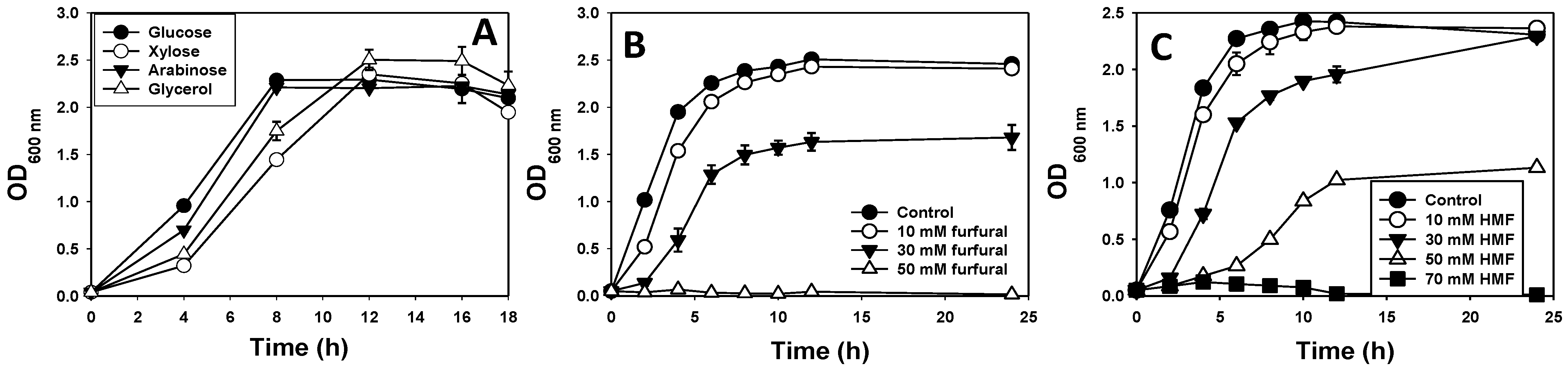
2.2. Assessment of Furfural and HMF Tolerance by E. hormaechei
2.3. Analytical Methods
2.4. DNA Extraction, Library Construction, and Whole-Genome Sequencing
2.5. Genome Assembly, Annotation and Gene Function Prediction
2.6. Phylogenetic Analysis, Digital DNA-DNA Hybridization (DDH) and Average Nucleotide Identity (ANI) Analyses
2.7. Genome Sequence Deposition and Data Availability
3. Results
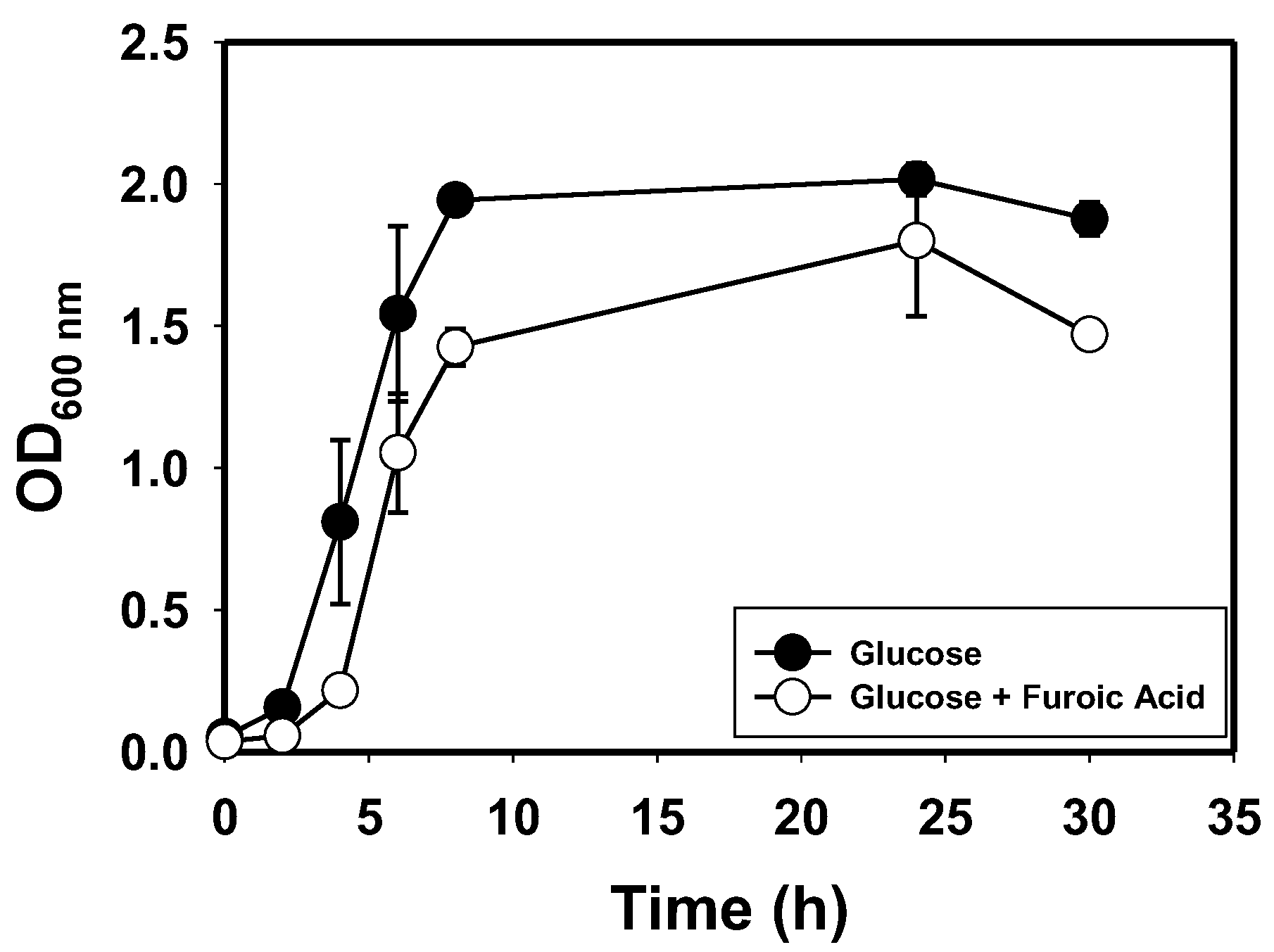
3.1. Species Identification, Growth Profiles and Furan Tolerance of E. hormaechei
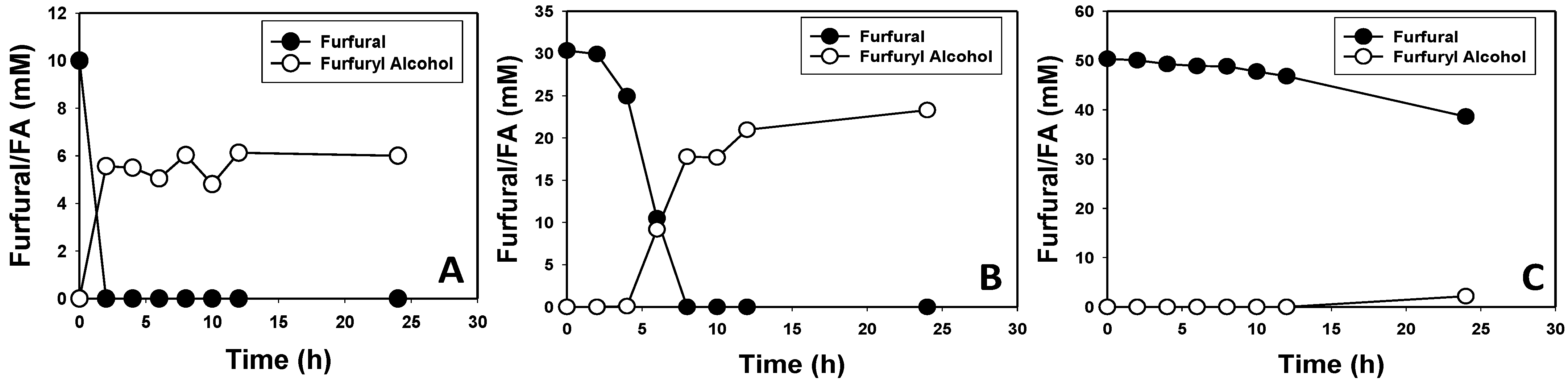
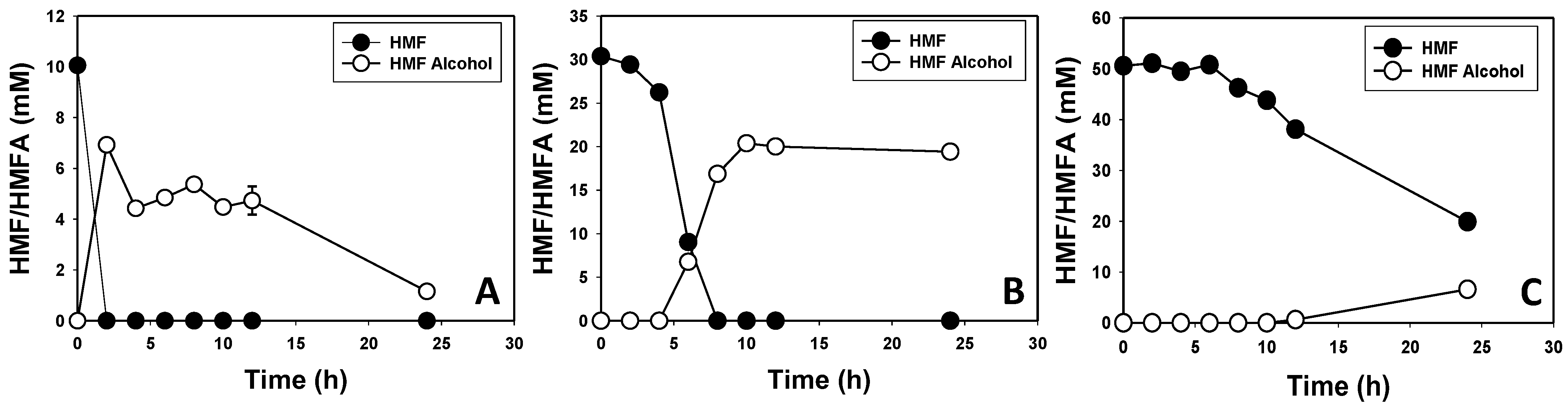
3.2. Furfural and HMF Are Largely Reduced to Their Less-Toxic Alcohols
3.3. Select metabolites of importance produced by E. hormaechei UW0SKVC1
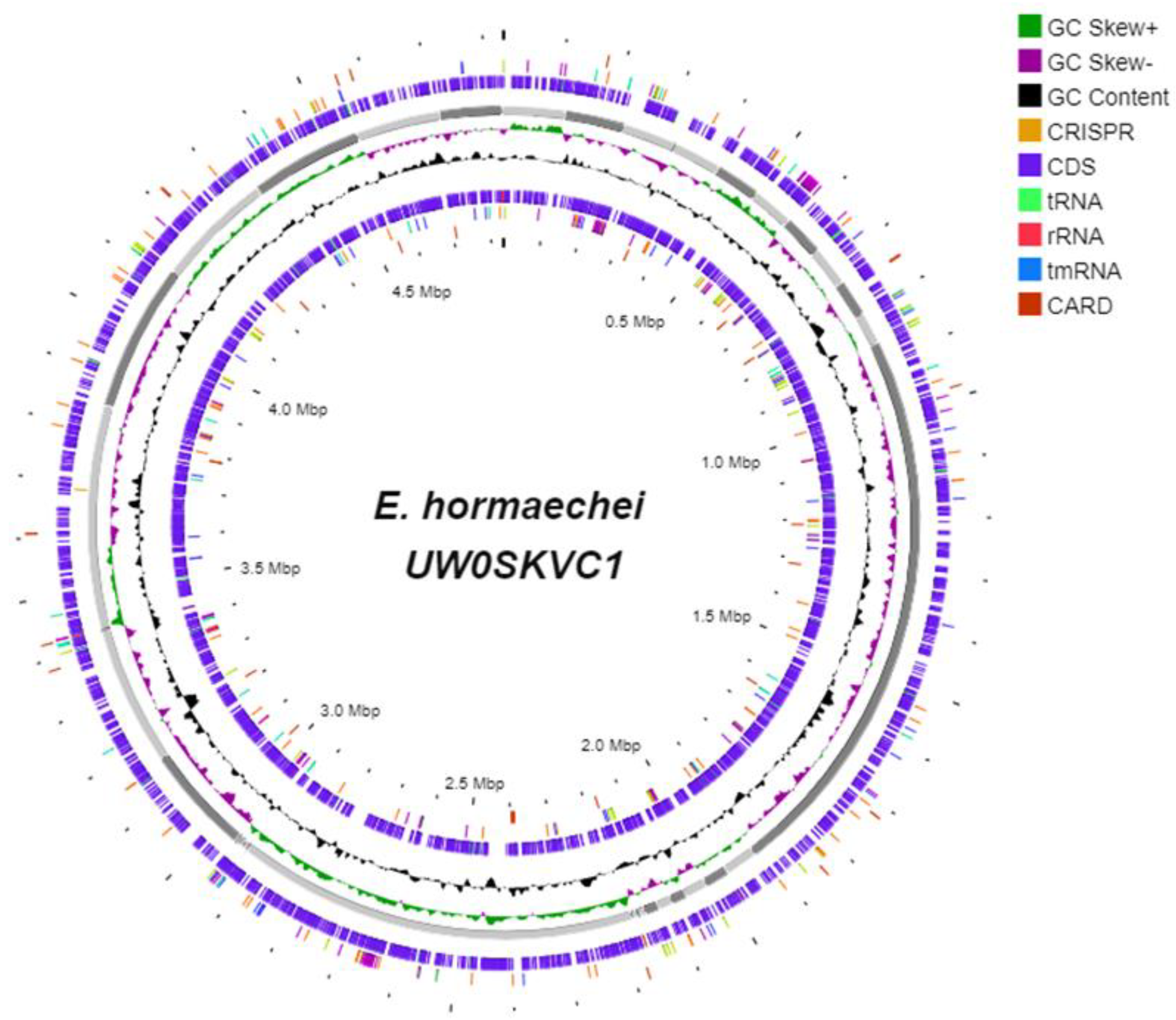
3.4. The Genomic Components of E. hormaechei UW0SKVC1 and Gene Annotations Analysis
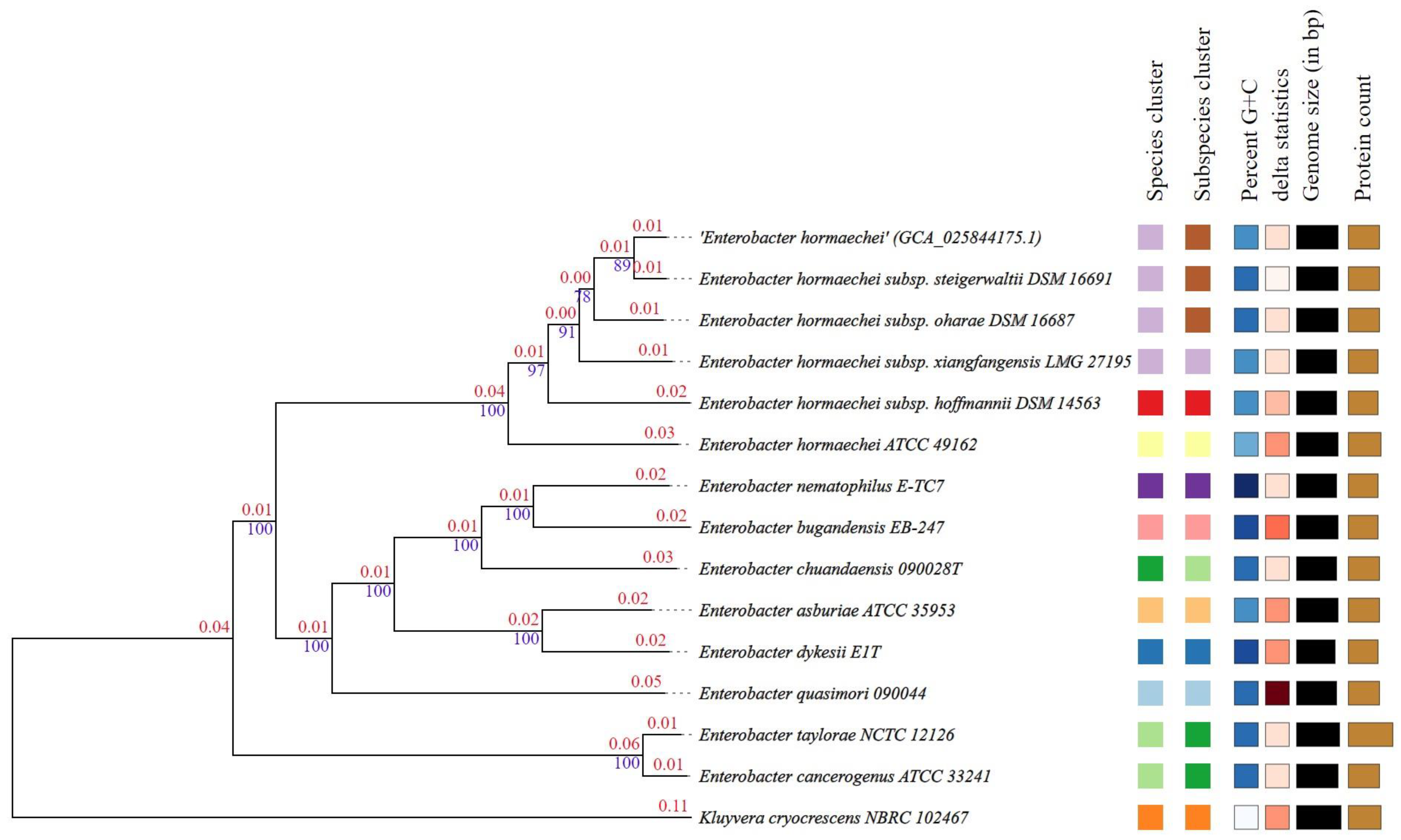
3.5. Phylogenetic Trees, Digital DDH and ANI Analysis
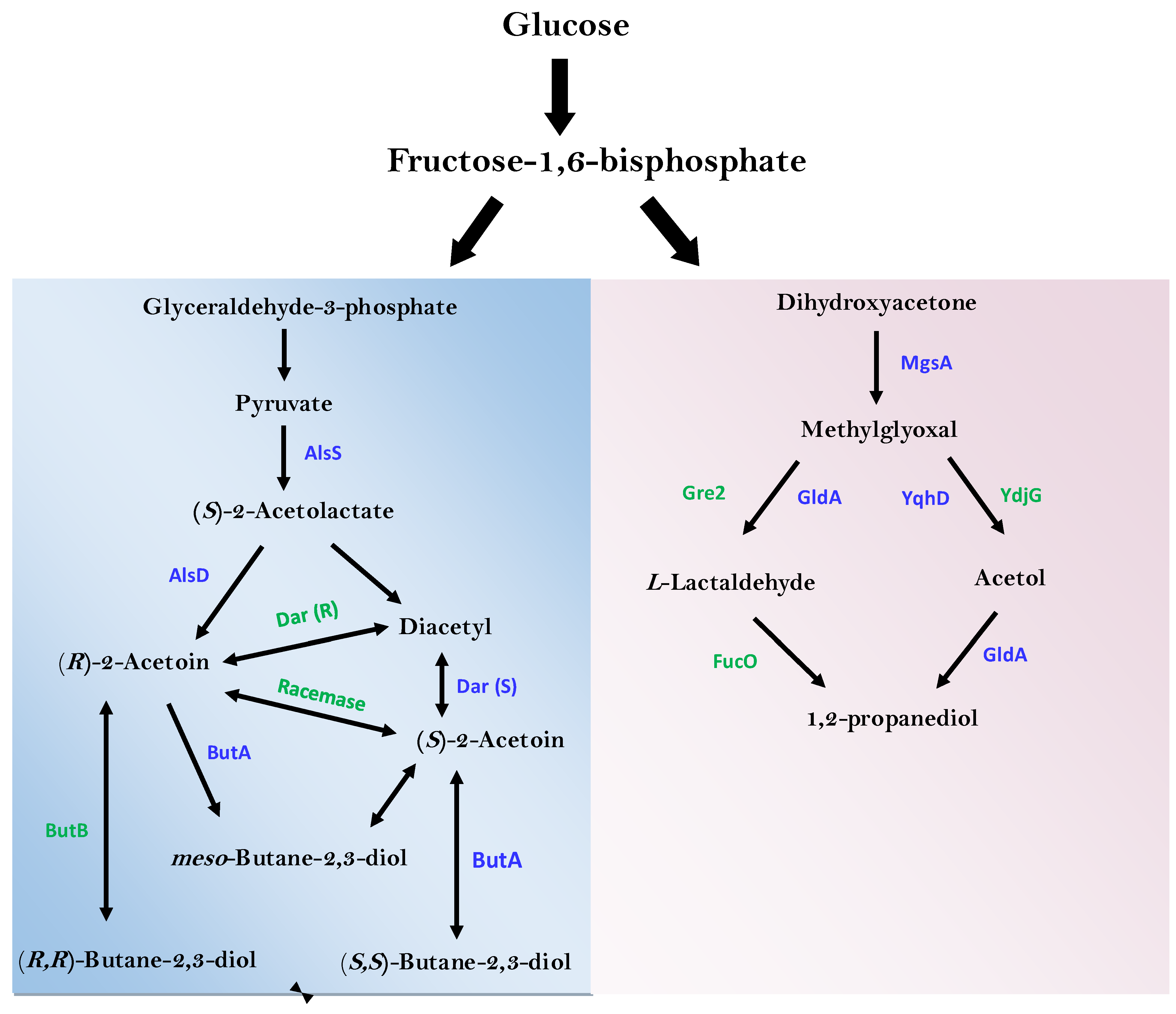
3.6. Identification of Enzymes of Industrial and Environmental Important
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chew, J.; Doshi, V. Recent advances in biomass pretreatment—Torrefaction fundamentals and technology. Renew. Sustain. Energy Rev. 2018, 15, 4212–4222. [Google Scholar] [CrossRef]
- Rosales-Calderon, O.; Arantes, V. A review on commercial-scale high-value products that can be produced alongside cellulosic ethanol. Biotechnol. Biofuels 2019, 12, 240. [Google Scholar] [PubMed]
- Guragain, Y.N.; Vadlani, P.V. Renewable biomass utilization: A way forward to establish sustainable chemical and processing industries. Clean Technol. 2021, 3, 243–259. [Google Scholar] [CrossRef]
- Mills, T.Y.; Sandoval, N.R.; Gill, R.T. Cellulosic hydrolysate toxicity and tolerance mechanisms in Escherichia coli. Biotechnol. Biofuels 2009, 2, 26. [Google Scholar] [CrossRef]
- Vanmarcke, G.; Demeke, M.M.; Foulquié-Moreno, M.R.; Thevelein, J.M. Identification of the major fermentation inhibitors of recombinant 2G yeasts in diverse lignocellulose hydrolysates. Biotechnol. Biofuels 2021, 14, 92. [Google Scholar] [CrossRef]
- Liu, K.; Atiyeh, H.K.; Pardo-Planas, O.; Ezeji, T.C.; Ujor, V.; Overton, J.C.; Berning, K.; Wilkins, M.R.; Tanner, R.S. Butanol production from hydrothermolysis-pretreated switchgrass: Quantification of inhibitors and detoxification of hydrolyzate. Bioresour. Technol. 2015, 189, 292–301. [Google Scholar] [CrossRef]
- Ujor, V.; Agu, C.V.; Gopalan, V.; Ezeji, T.C. Glycerol supplementation of the growth medium enhances in situ detoxification of furfural by Clostridium beijerinckii during butanol fermentation. Appl. Microbiol. Biotechnol. 2014, 98, 6511–6521. [Google Scholar] [CrossRef]
- Palmqvist, E.; Almeida, J.S.; Hahn-Hägerdal, B. Influence of furfural on anaerobic glycolytic kinetics of Saccharomyces cerevisiae in batch culture. Biotechnol. Bioeng. 1999, 62, 447–454. [Google Scholar] [CrossRef]
- Almeida, J.R.; Röder, A.; Modig, T.; Laadan, B.; Lidén, G.; Gorwa-Grauslund, M.F. NADH- vs. NADPH-coupled reduction of 5-hydroxymethyl furfural (HMF) and its implications on product distribution in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2008, 78, 939–945. [Google Scholar] [CrossRef]
- Richmond, C.; Ujor, V.; Ezeji, T.C. Impact of syringaldehyde on the growth of Clostridium beijerinckii NCIMB 8052 and butanol production. 3 Biotech 2012, 2, 159–167. [Google Scholar] [CrossRef][Green Version]
- Yan, Y.; Bu, C.; He, Q.; Zheng, Z.; Ouyang, J. Efficient bioconversion of furfural to furfuryl alcohol by Bacillus coagulans NL01. RSC Adv. 2018, 8, 26720–26727. [Google Scholar] [CrossRef]
- Lu, P.; Chen, L.J.; Li, G.X.; Shen, S.H.; Wang, L.L.; Jiang, Q.Y.; Zhang, J.F. Influence of furfural concentration on growth and ethanol yield of Saccharomyces kluyveri. J. Environ. Sci. 2007, 19, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Deng, C.; Cheng, J.; Murphy, J.D. Low concentrations of furfural facilitate biohydrogen production in dark fermentation using Enterobacter aerogenes. Renew. Energy 2020, 150, 23–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, B.; Ezeji, T.C. Biotransformation of furfural and 5-hydroxymethyl furfural (HMF) by Clostridium acetobutylicum ATCC 824 during butanol fermentation. New Biotechnol. 2012, 29, 345–351. [Google Scholar] [CrossRef]
- Koopman, F.; Wierckx, N.; de Winde, J.H.; Ruijssenaars, H.J. Identification and characterization of the furfural and 5-(hydroxymethyl) furfural degradation pathways of Cupriavidus basilensis HMF14. Proc. Natl. Acad. Sci. USA 2010, 107, 4919–4924. [Google Scholar] [CrossRef]
- Wierckx, N.; Koopman, F.; Ruijssenaars, H.J.; de Winde, J.H. Microbial degradation of furanic compounds: Biochemistry, genetics, and impact. Appl. Microbiol. Biotechnol. 2011, 92, 1095–1105. [Google Scholar] [CrossRef]
- Crigler, J.; Eiteman, M.A.; Altman, E. Characterization of the Furfural and 5-Hydroxymethylfurfural (HMF) Metabolic Pathway in the Novel Isolate Pseudomonas putida ALS1267. Appl. Biochem. Biotechnol. 2019, 190, 918–930. [Google Scholar] [CrossRef]
- Miller, E.N.; Jarboe, L.R.; Turner, P.C.; Pharkya, P.; Yomano, L.P.; York, S.W.; Nunn, D.; Shanmugam, K.T.; Ingram, L.O. Furfural inhibits growth by limiting sulfur assimilation in ethanologenic Escherichia coli strain LY180. Appl. Environ. Microbiol. 2009, 75, 6132–6141. [Google Scholar] [CrossRef]
- Miller, E.N.; Jarboe, L.R.; Yomano, L.P.; York, S.W.; Shanmugam, K.T.; Ingram, L.O. Silencing of NADPH-dependent oxidoreductase genes (yqhD and dkgA) in furfural-resistant ethanologenic Escherichia coli. Appl. Environ. Microbiol. 2009, 75, 4315–4323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wang, X.; Yomano, L.; Shanmugam, K.; Ingram, L. Increase in Furfural Tolerance in Ethanologenic Escherichia coli LY180 by Plasmid-Based Expression of thyA. Appl. Environ. Microbiol. 2012, 78, 4346–4352. [Google Scholar] [CrossRef]
- Willson, B.J.; Herman, R.; Langer, S.; Thomas, G.H. Improved furfural tolerance in Escherichia coli mediated by heterologous NADH-dependent benzyl alcohol dehydrogenases. Biochem. J. 2022, 479, 1045–1058. [Google Scholar] [CrossRef]
- Wang, X.; Yomano, L.P.; Lee, J.Y.; York, S.W.; Zheng, H.; Mullinnix, M.T.; Shanmugam, K.T.; Ingram, L.O. Engineering furfural tolerance in Escherichia coli improves the fermentation of lignocellulosic sugars into renewable chemicals. Proc. Natl. Acad. Sci. USA 2012, 110, 4021–4026. [Google Scholar] [CrossRef]
- Okonkwo, C.C.; Ujor, V.; Ezeji, T.C. Chromosomal integration of aldo-keto-reductase and short-chain dehydrogenase/reductase genes in Clostridium beijerinckii NCIMB 8052 enhanced tolerance to lignocellulose-derived microbial inhibitory compounds. Sci. Rep. 2019, 9, 7634. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Kong, S.; Luo, S.; Chen, C.; Cui, Z.; Sun, X.; Chen, T.; Wang, Z. Improving Furfural tolerance of Escherichia coli by integrating adaptive laboratory evolution with CRISPR-enabled trackable genome engineering (CREATE). ACS Sustain. Chem. Eng. 2022, 10, 2318–2330. [Google Scholar] [CrossRef]
- Choi, S.Y.; Gong, G.; Park, H.S.; Um, Y.; Sim, S.J.; Woo, H.M. Extreme furfural tolerance of a soil bacterium Enterobacter cloacae GGT036. J. Biotechnol. 2015, 193, 11–13. [Google Scholar] [CrossRef]
- Boopathy, R.; Bokang, H.; Daniels, L. Biotransformation of furfural and 5-hydroxymethyl furfural by enteric bacteria. J. Ind. Microbiol. 1993, 11, 147–150. [Google Scholar] [CrossRef]
- Zhang, D.; Ong, Y.L.; Li, Z.; Wu, J.C. Biological detoxification of furfural and 5-hydroxyl methyl furfural in hydrolysate of oil palm empty fruit bunch by Enterobacter sp. FDS8. Biochem. Eng. J. 2013, 72, 77–82. [Google Scholar] [CrossRef]
- Agyeman-Duah, E.; Kumar, S.; Gangwar, B.; Ujor, V.C. Glycerol utilization as a sole carbon source disrupts the membrane architecture and solventogenesis in Clostridium beijerinckii NCIMB 8052. Fermentation 2022, 8, 339. [Google Scholar] [CrossRef]
- Kumar, S.; Ujor, V.C. Complete genome sequence of Paenibacillus polymyxa DSM 365, a soil bacterium of agricultural and industrial importance. Microbiol. Resour. 2022, 11, e0032922. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.; Bolund, L.; Wang, J. State of the art de novo assembly of human genomes from massively parallel sequencing data. Hum. Genom. 2010, 4, 271. [Google Scholar] [CrossRef][Green Version]
- Lin, S.H.; Liao, Y.C. CISA: Contig integrator for sequence assembly of bacterial genomes. PLoS ONE 2013, 8, e60843. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Sardà Carbasse, J.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acid Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–667. [Google Scholar] [CrossRef]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2014, 110, 1281–1286. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.; Murray, R.; Stackebrandt, E.S.; et al. Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Chun, J.; Rainey, F.A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64, 316–324. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Zaldivar, J.; Martinez, A.; Ingram, L.O. Effect of selected aldehydes on the growth and fermentation of ethanologenic Escherichia coli. Biotechnol. Bioeng. 1999, 65, 24–33. [Google Scholar] [CrossRef]
- Zhang, Y.; Ezeji, T.C. Transcriptional analysis of Clostridium beijerinckii NCIMB 8052 to elucidate role of furfural stress during acetone butanol ethanol fermentation. Biotechnol. Biofuels 2013, 6, 66. [Google Scholar] [CrossRef]
- Zhang, Y.; Ezeji, T.C. Elucidating and alleviating impacts of lignocellulose-derived microbial inhibitors on Clostridium beijerinckii during fermentation of Miscanthus giganteus to butanol. J. Ind. Microbiol. Biotechnol. 2014, 41, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Field, S.J.; Ryden, P.; Wilson, D.; James, S.A.; Roberts, I.N.; Richardson, D.J.; Waldron, K.W.; Clarke, T.A. Identification of furfural resistant strains of Saccharomyces cerevisiae and Saccharomyces paradoxus from a collection of environmental and industrial isolates. Biotechnol. Biofuels 2015, 8, 33. [Google Scholar] [CrossRef]
- Allen, S.A.; Clark, W.; McCaffery, J.M.; Cai, Z.; Lanctot, A.; Slininger, P.J.; Liu, Z.L.; Gorsich, S.W. Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol. Biofuels 2010, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Um, Y.; Park, T.H.; Woo, H.M. Complete genome sequence of Enterobacter cloacae GGT036: A furfural tolerant soil bacterium. J. Biotechnol. 2015, 193, 43–44. [Google Scholar] [CrossRef]
- Degelmann, D.M.; Kolb, S.; Dumont, M.; Murrell, J.C.; Drake, H.L. Enterobacteriaceae facilitate the anaerobic degradation of glucose by a forest soil. FEMS Microbiol. Ecol. 2009, 68, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.M. Naturally Occurring Ring Compounds; Butterworths: London, UK, 1963; pp. 1–15. [Google Scholar]
- Trudgill, P.W. The microbial metabolism of furans. In Microbial Degradation of Organic Compounds; Gibson, D.T., Ed.; Marcel Dekker: New York, NY, USA, 1984; pp. 295–307. [Google Scholar]
- Okonkwo, C.C.; Ujor, V.; Ezeji, T.C. Production of 2,3-Butanediol from non-detoxified wheat straw hydrolysate: Impact of microbial inhibitors on Paenibacillus polymyxa DSM 365. Ind. Crops Prod. 2021, 159, 113047. [Google Scholar] [CrossRef]
- Xiu, Z.L.; Zeng, A.P. Present state and perspective of downstream processing of biologically produced 1,3-propanediol and 2,3-butanediol. Appl. Microbiol. Biotechnol. 2008, 78, 917–926. [Google Scholar] [CrossRef]
- Köpke, M.; Mihalcea, C.; Liew, F.; Tizard, J.H.; Ali, M.S.; Conolly, J.J.; Al-Sinawi, B.; Simpson, S.D. 2,3-butanediol production by acetogenic bacteria, an alternative route to chemical synthesis, using industrial waste gas. Appl. Environ. Microbiol. 2011, 77, 5467–5475. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Wang, Z.; Zheng, M.; Chen, T. Advances in biological production of acetoin: A comprehensive overview. Crit. Rev. Biotechnol. 2022, 42, 1135–1156. [Google Scholar] [CrossRef]
- Wu, Y.; Chu, W.; Yang, J.; Xu, Y.; Shen, Q.; Yang, H.; Xu, F.; Liu, Y.; Lu, P.; Jiang, K.; et al. Metabolic Engineering of Enterobacter aerogenes for Improved 2,3-Butanediol Production by Manipulating NADH Levels and Overexpressing the Small RNA RyhB. Front. Microbiol. 2021, 12, 754306. [Google Scholar] [CrossRef] [PubMed]
- Amraoui, Y.; Prabhu, A.A.; Narisetty, V.; Coulon, F.; Chandel, A.K.; Willoughby, N.; Jacob, S.; Koutinas, A.; Kumar, V. Enhanced 2,3-Butanediol production by mutant Enterobacter ludwigii using Brewers’ spent grain hydrolysate: Process optimization for a pragmatic biorefinery loom. Chem. Eng. J. 2022, 427, 130851. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Peng, X.P.; Li, W.; Guo, X.W.; Xiao, D.G. Optimization of 2,3-butanediol production by Enterobacter cloacae in simultaneous saccharification and fermentation of corncob residue. Biotechnol. Appl. Biochem. 2014, 61, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.C.; Bothast, R.J. Production of 2,3-butanediol by newly isolated Enterobacter cloacae. Appl. Microbiol. Biotechnol. 1999, 52, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.K.; Anand, P.; Saran, S.; Isar, J.; Agarwal, L. Microbial production and applications of 1,2-propanediol. Indian J. Microbiol. 2010, 50, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, H.; Kashket, S.; Young, M.; Kashket, E.R. Clostridium beijerinckii and Clostridium difficile detoxify methylglyoxal by a novel mechanism involving glycerol dehydrogenase. Appl. Environ. Microbiol. 2001, 67, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-c.; Dend, Y.-l.; Wang, H.-y.; Wang, C.-g.; Ma, L.-l.; Liu, Q.-y. Production of acetol and lactic acid from cellulose hydrogenolysis over Sn-Fe@C catalysts. J. Fuel Chem. Technol. 2022, 50, 314–325. [Google Scholar] [CrossRef]
- Cheon, Y.; Kim, J.S.; Park, J.B.; Heo, P.; Lim, J.H.; Jung, G.Y.; Seo, J.H.; Park, J.H.; Koo, H.M.; Cho, K.M.; et al. A biosynthetic pathway for hexanoic acid production in Kluyveromyces marxianus. J. Biotechnol. 2014, 182–183, 30–36. [Google Scholar] [CrossRef]
- Goldberg, I.; Rokem, J.S. Organic and Fatty Acid Production, Microbial. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 421–442. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified Compounds | * Mineral Medium | * Rich Medium | ||||||
|---|---|---|---|---|---|---|---|---|
| Aerobic | Anaerobic | Aerobic | Anaerobic | |||||
| 24 h | 60 h | 24 h | 60 h | 24 h | 60 h | 24 h | 60 h | |
| 2,3-Butanediol | + | + | + | + | + | + | + | + |
| 1,2-Cyclopentanedione | + | + | - | + | - | - | - | - |
| Acetoin (2-Butanone) | + | - | + | - | + | + | + | + |
| Ethanol | - | + | - | - | + | - | - | - |
| Acetic acid | + | + | + | + | + | + | + | + |
| Formic acid | - | - | - | + | + | - | + | + |
| Acetol (hydroxyacetone) | + | - | + | + | - | - | + | - |
| Hexanoic acid | + | + | + | + | - | - | - | - |
| Pentanoic acid | - | + | + | - | - | - | - | - |
| L-lactic acid | - | - | - | - | - | + | + | + |
| 2,3-BD concentrations (g/L) | ||||||||
| Aerobic | Anaerobic | |||||||
| Glucose | 42.64 ± 0.75 | 4.60 ± 0.2 | ||||||
| Lactose | 41.65 ± 0.93 | 3.39 ± 0.097 | ||||||
| Functional Annotation Against Databases (Protein Coding Genes) | Non-Redundant ncRNAs | Interspersed Repeats | Tandem Repeats | ||||
|---|---|---|---|---|---|---|---|
| nr KEGG SwissProt COG GO Pfam | 4540 4508 3483 3845 3276 3276 | tRNA 5 s (denovo) 16 s (denovo) 23 s (denovo) sRNA | 78 8 1 1 41 | LTR DNA LINE SINE RE Unknown | 36 12 12 17 0 0 | TR Minisatellite DNA Microsatellite DNA | 78 52 1 |
| Biological Pathway Type | No. of Genes | Biological Pathway Type | No. of Genes |
|---|---|---|---|
| Cellular Processes Transport and catabolism Cell growth and death Cell motility Cellular community—prokaryotes | 8 177 69 22 | Metabolism Xenobiotics biodegradation and metabolism Nucleotide metabolism Metabolism of terpenoids and polyketides Amino acids metabolism Metabolism of cofactors and vitamins Lipid metabolism Glycan biosynthesis and metabolism Energy metabolism Carbohydrate metabolism Biosynthesis of other secondary metabolites (e.g., siderophores) | 37 105 33 298 182 66 57 164 340 36 |
| Environmental Information Processing Signal transduction Membrane transport | 169 329 | ||
| Genetic Information Processing Translation Transcription Replication and repair Folding, sorting and degradation | 80 4 56 53 | ||
| Human Diseases Neurodegenerative diseases Infectious diseases Immune diseases Endocrine and metabolic diseases Drug resistance Cardiovascular diseases Cancers | 6 40 2 6 72 7 18 | Organismal Systems Nervous system Immune system Excretory system Environmental adaptation Endocrine system Digestive system Aging | 2 4 1 8 12 2 9 |
| Query Strain | Subject Strain | dDDH (d0, %) | C.I. (d0, %) | dDDH (d4, %) | C.I. (d4, %) | dDDH (d6, %) | C.I. (d6, %) | G + C Difference (%) |
|---|---|---|---|---|---|---|---|---|
| E. hormaechei UW0SKVC1 | Enterobacter hormaechei subsp. steigerwaltii DSM 16691 | 87.3 | [83.7–90.1] | 91.4 | [89.2–93.1] | 90.7 | [88.0–92.8] | 0.19 |
| Enterobacter hormaechei subsp. oharae DSM 16687 | 85.1 | [81.4–88.2] | 80.6 | [77.7–83.2] | 87.3 | [84.2–89.8] | 0.22 | |
| Enterobacter hormaechei subsp. xiangfangensis LMG 27195 | 81.8 | [77.9–85.1] | 76.1 | [73.1–78.9] | 83.7 | [80.5–86.5] | 0.08 | |
| Enterobacter hormaechei subsp. hoffmannii DSM 14563 | 82.3 | [78.4–85.6] | 66.3 | [63.3–69.1] | 82.1 | [78.8–85.0] | 0.02 | |
| Enterobacter hormaechei ATCC 49162 | 77.4 | [73.4–80.9] | 61.3 | [58.4–64.1] | 76.8 | [73.3–79.9] | 0.12 | |
| Enterobacter bugandensis EB-247 | 71.8 | [67.8–75.4] | 35.5 | [33.0–38.0] | 62.7 | [59.4–65.9] | 0.64 | |
| Enterobacter quasimori 090044 | 73.2 | [69.3–76.9] | 35 | [32.6–37.5] | 63.5 | [60.2–66.8] | 0.4 | |
| Enterobacter chuandaensis 090028T | 69 | [65.1–72.7] | 35 | [32.5–37.5] | 60.4 | [57.1–63.6] | 0.32 | |
| Enterobacter asburiae ATCC 35953 | 61 | [47.4–64.6] | 34.6 | [32.2–37.1] | 54.3 | [51.2–57.4] | 0.11 | |
| Enterobacter chengduensis WCHECl-C4 | 61.4 | [57.7–64.9] | 34.1 | [31.7–36.6] | 54.3 | [51.2–57.4] | 0.38 | |
| Enterobacter dykesii E1T | 67.2 | [63.4–70.9] | 33.8 | [31.4–36.3] | 58.5 | [55.3–61.6] | 0.49 | |
| Enterobacter taylorae NCTC 12126 | 62.7 | [59.0–66.3] | 31.6 | [29.2–34.1] | 54.1 | [50.9–57.2] | 0.37 | |
| Enterobacter cancerogenus ATCC 33241 | 66.1 | [62.3–69.8] | 31.4 | [29.0–33.9] | 56.3 | [53.2–59.4] | 0.32 |
| E.C. Number | Enzyme Symbol | Enzyme Name | Status in E. hormaechei |
|---|---|---|---|
| 2.2.16 | AlsS | Acetolactate synthase | Present |
| 4.1.1.5 | AlsD | Acetolactate decarboxylase | Present |
| 1.1.1.303 | Dar (R) | Diacetyl reductase | Absent |
| 1.1.1.304 | Dar (S) | Diacetyl reductase | Present |
| 5.1.2.4 | Acetoin racemase | Absent | |
| 1.1.1.4 | ButB | (R,R)-Butanediol dehydrogenase | Absent |
| 1.1.1.- | ButA | meso-Butanediol dehydrogenase | Present |
| 1.1.1.76 | ButA | (S,S)-Butanediol dehydrogenase | Present |
| 4.2.3.3 | MgsA | Methylglyoxal synthase | Present |
| 1.1.-.- | YqhD | NADP-dependent alcohol dehydrogenase | Present |
| 1.1.1.- | YdjG | Methylglyoxal reductase | Absent |
| 1.1.1.6 | GldA | Glycerol dehydrogenase | Present |
| 1.1.1.283 | Gre2 | NADPH-dependent methylglyoxal reductase | Absent |
| 1.1.1.77 | FucO | Lactaldehyde reductase | Absent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Agyeman-Duah, E.; Ujor, V.C. Whole-Genome Sequence and Fermentation Characteristics of Enterobacter hormaechei UW0SKVC1: A Promising Candidate for Detoxification of Lignocellulosic Biomass Hydrolysates and Production of Value-Added Chemicals. Bioengineering 2023, 10, 1090. https://doi.org/10.3390/bioengineering10091090
Kumar S, Agyeman-Duah E, Ujor VC. Whole-Genome Sequence and Fermentation Characteristics of Enterobacter hormaechei UW0SKVC1: A Promising Candidate for Detoxification of Lignocellulosic Biomass Hydrolysates and Production of Value-Added Chemicals. Bioengineering. 2023; 10(9):1090. https://doi.org/10.3390/bioengineering10091090
Chicago/Turabian StyleKumar, Santosh, Eric Agyeman-Duah, and Victor C. Ujor. 2023. "Whole-Genome Sequence and Fermentation Characteristics of Enterobacter hormaechei UW0SKVC1: A Promising Candidate for Detoxification of Lignocellulosic Biomass Hydrolysates and Production of Value-Added Chemicals" Bioengineering 10, no. 9: 1090. https://doi.org/10.3390/bioengineering10091090
APA StyleKumar, S., Agyeman-Duah, E., & Ujor, V. C. (2023). Whole-Genome Sequence and Fermentation Characteristics of Enterobacter hormaechei UW0SKVC1: A Promising Candidate for Detoxification of Lignocellulosic Biomass Hydrolysates and Production of Value-Added Chemicals. Bioengineering, 10(9), 1090. https://doi.org/10.3390/bioengineering10091090