
CycP: A Novel Self-Assembled Vesicle-Forming Cyclic Antimicrobial Peptide to Control Drug-Resistant S. aureus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Solid-Phase Peptide Synthesis, Purification, and Molecular Weight Determination
2.2. Multiple Sequence Alignment and De Novo Peptide Structure Prediction
2.3. Bacterial Strain and Media
2.4. Peptide Self-Assembly and Vesicle Preparation
2.5. Minimum Inhibitory Concentration Determination
2.6. SEM Sample Preparation for Peptide Vesicles
2.7. TEM Sample Preparation for Bacterial Cells
2.8. Molecular Docking
2.9. Cell Toxicity Assay
2.10. Statistical Analysis
3. Results
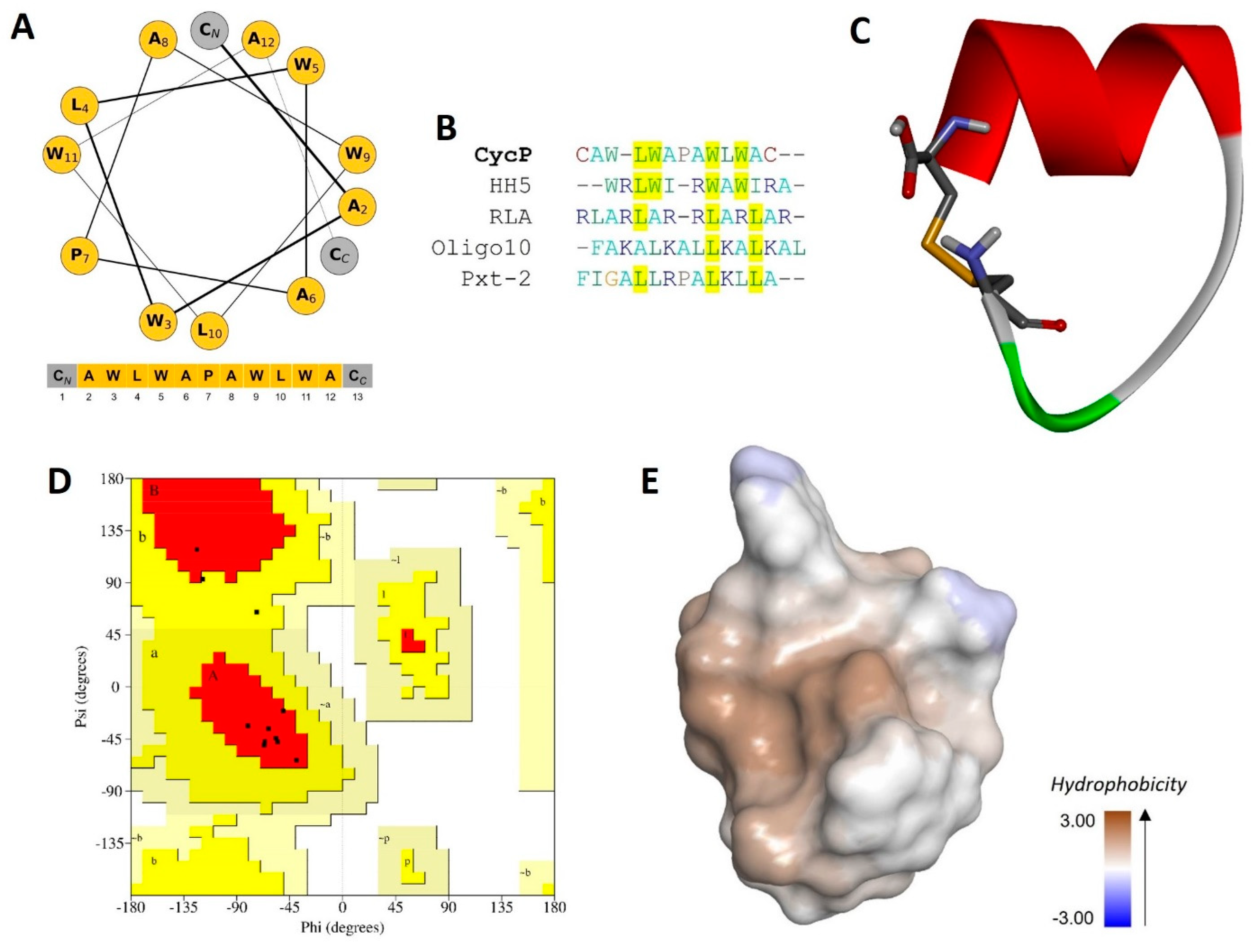
3.1. CycP Is a Novel Cyclic Peptide
3.2. CycP Forms Self-Assembled Peptide Vesicles
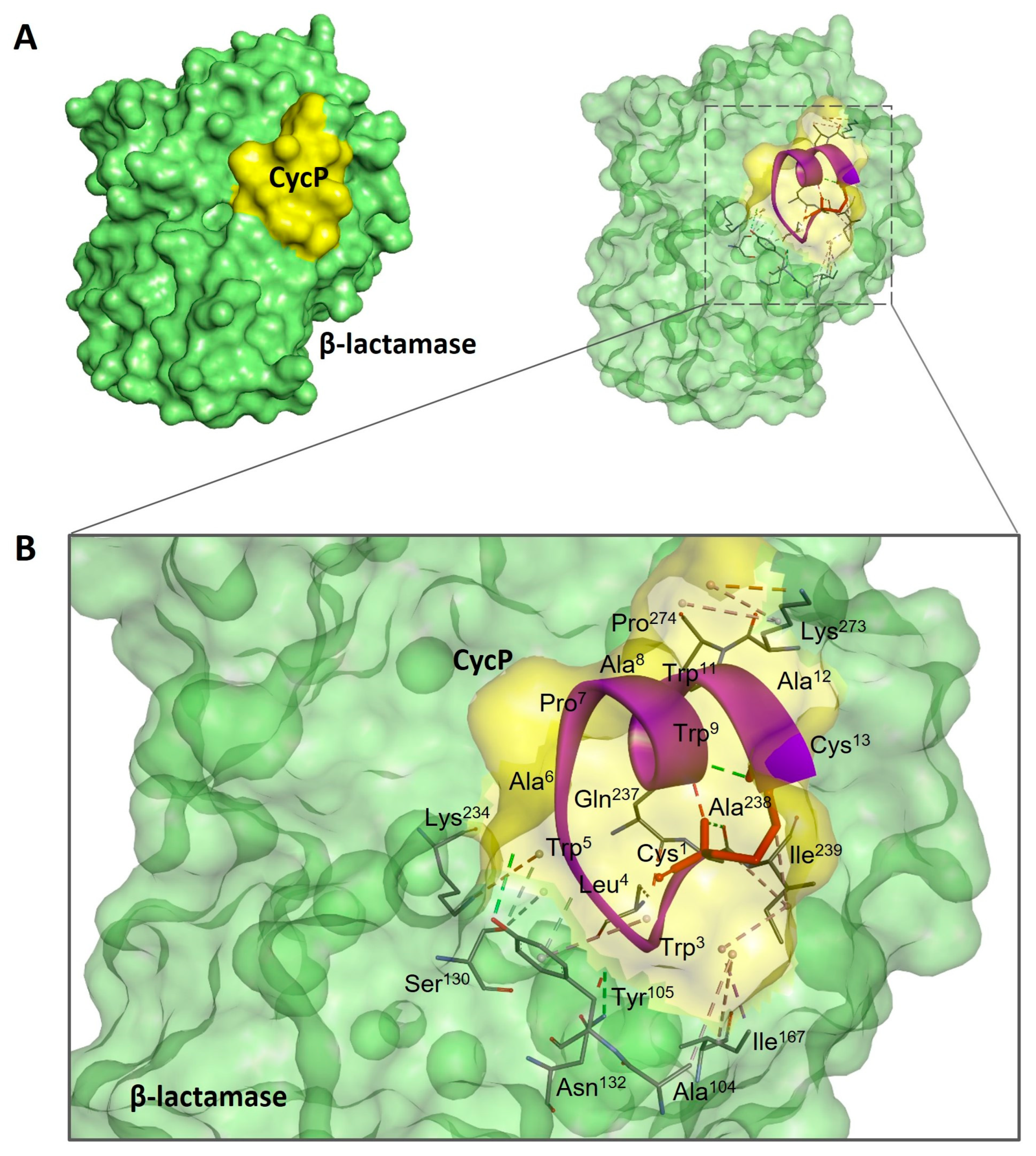
3.3. CycP Is a Dual-Action Cyclic AMP
3.4. Self-Assembled CycP Vesicles Deliver the Antibiotic and Kill the Drug-Resistant S. aureus in a Membrane-Specific Mechanism of Action
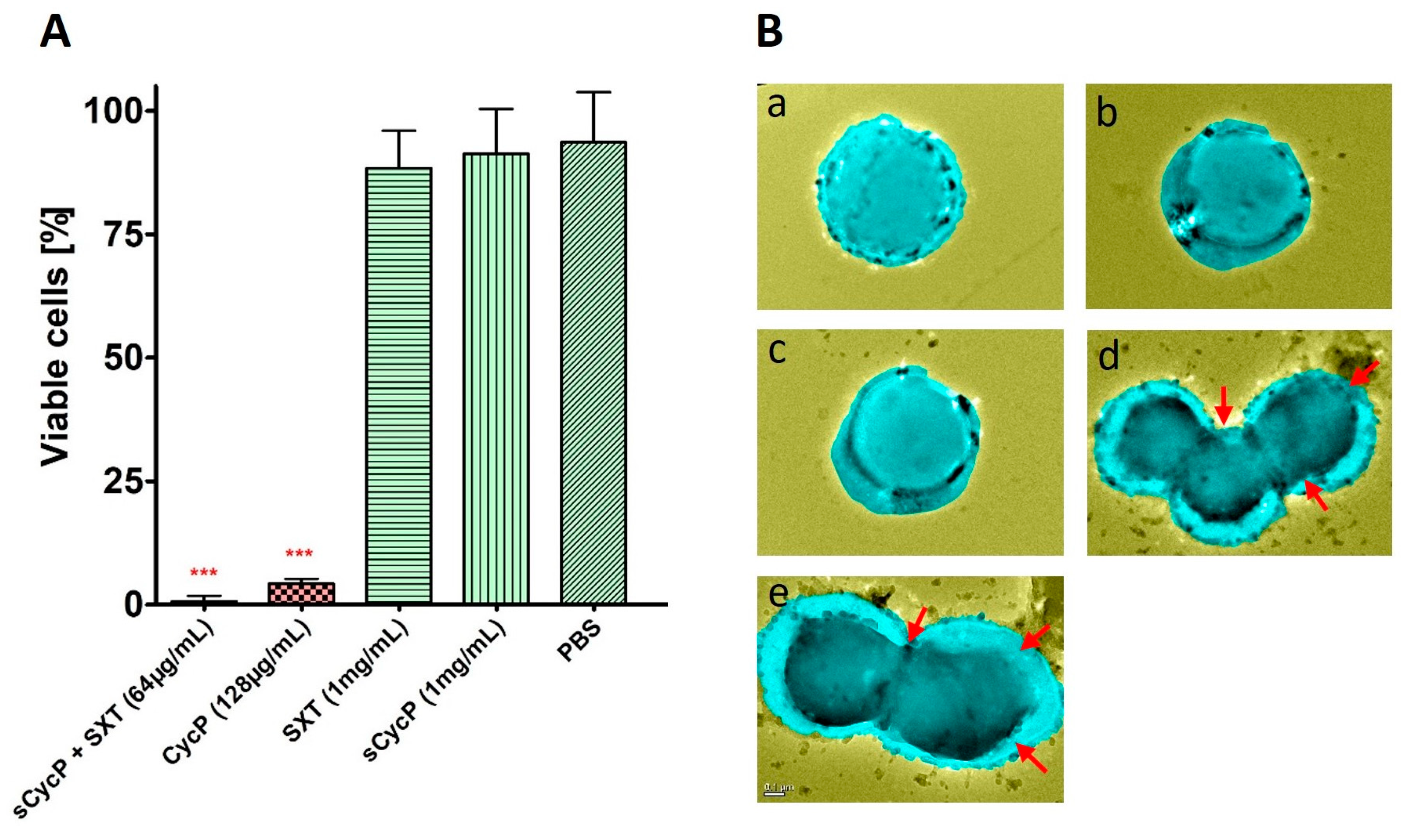
3.5. CycP and sCycP Vesicles are Non-Cytotoxic
4. Discussion
5. Conclusions
6. Limitations of the Present Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xuan, J.; Feng, W.; Wang, J.; Wang, R.; Zhang, B.; Bo, L.; Chen, Z.S.; Yang, H.; Sun, L. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist. Updat. 2023, 68, 100954. [Google Scholar]
- Fadaka, A.O.; Sibuyi, N.R.S.; Madiehe, A.M.; Meyer, M. Nanotechnology-based delivery systems for antimicrobial peptides. Pharmaceutics 2021, 13, 1795. [Google Scholar] [CrossRef]
- Dehsorkhi, A.; Castelletto, V.; Hamley, I.W. Self-assembling amphiphilic peptides. J. Pept. Sci. 2014, 20, 453–467. [Google Scholar]
- Tarvirdipour, S.; Schoenenberger, C.A.; Benenson, Y.; Palivan, C.G. A self-assembling amphiphilic peptide nanoparticle for the efficient entrapment of DNA cargoes up to 100 nucleotides in length. Soft Matter 2020, 16, 1678–1691. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, H.; Huang, Y.; Lian, B.; Ma, C.; Han, L.; Chen, Y.; Wu, S.; Li, N.; Zhang, W.; et al. A self-assembling amphiphilic peptide dendrimer-based drug delivery system for cancer therapy. Pharmaceutics 2021, 13, 1092. [Google Scholar] [CrossRef]
- Lee, S.; Trinh, T.H.T.; Yoo, M.; Shin, J.; Lee, H.; Kim, J.; Hwang, E.; Lim, Y.B.; Ryou, C. Self-assembling peptides and their application in the treatment of diseases. Int. J. Mol. Sci. 2019, 20, 5850. [Google Scholar] [CrossRef]
- Gudlur, S.; Sukthankar, P.; Gao, J.; Avila, L.A.; Hiromasa, Y.; Chen, J.; Iwamoto, T.; Tomich, J.M. Peptide Nanovesicles Formed by the Self-Assembly of Branched Amphiphilic Peptides. PLoS ONE 2012, 7, e45374. [Google Scholar] [CrossRef]
- Schnaider, L.; Brahmachari, S.; Schmidt, N.W.; Mensa, B.; Shaham-Niv, S.; Bychenko, D.; Adler-Abramovich, L.; Shimon, L.J.W.; Kolusheva, S.; Degrado, W.F.; et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 2017, 8, 1365. [Google Scholar] [CrossRef]
- Pandit, G.; Roy, K.; Agarwal, U.; Chatterjee, S. Self-Assembly Mechanism of a Peptide-Based Drug Delivery Vehicle. ACS Omega 2018, 3, 3143–3155. [Google Scholar] [CrossRef]
- Mahata, D.; Mandal, S.M.; Bharti, R.; Gupta, V.K.; Mandal, M.; Nag, A.; Nando, G.B. Self-assembled cardanol azo derivatives as antifungal agent with chitin-binding ability. Int. J. Biol. Macromol. 2014, 69, 5–11. [Google Scholar] [CrossRef]
- Fujita, S.; Motoda, Y.; Kigawa, T.; Tsuchiya, K.; Numata, K. Peptide-Based Polyion Complex Vesicles That Deliver Enzymes into Intact Plants to Provide Antibiotic Resistance without Genetic Modification. Biomacromolecules 2021, 22, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Lee, D.; Kim, H.; Jung, Y.; Koo, H.; Lim, Y. beom Structural control of self-assembled peptide nanostructures to develop peptide vesicles for photodynamic therapy of cancer. Mater. Today Bio 2022, 16, 100337. [Google Scholar] [CrossRef]
- Mandal, S.M.; Migliolo, L.; Silva, O.N.; Fensterseifer, I.C.M.; Faria-Junior, C.; Dias, S.C.; Basak, A.; Hazra, T.K.; Franco, O.L. Controlling resistant bacteria with a novel class of β 2-lactamase inhibitor peptides: From rational design to in vivo analyses. Sci. Rep. 2014, 4, srep06015. [Google Scholar] [CrossRef]
- Baindara, P.; Chaudhry, V.; Mittal, G.; Liao, L.M.; Matos, C.O.; Khatri, N.; Franco, O.L.; Patil, P.B.; Korpole, S. Characterization of the antimicrobial peptide penisin, a class Ia novel lantibiotic from Paenibacillus sp. strain A3. Antimicrob. Agents Chemother. 2016, 60, 580–591. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Sellers, B.D.; Zhu, K.; Zhao, S.; Friesner, R.A.; Jacobson, M.P. Toward better refinement of comparative models: Predicting loops in inexact environments. Proteins Struct. Funct. Genet. 2008, 72, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M.; Khan, J.; Mahata, D.; Saha, S.; Sengupta, J.; Silva, O.N.; Das, S.; Mandal, M.; Franco, O.L. A self-assembled clavanin A-coated amniotic membrane scaffold for the prevention of biofilm formation by ocular surface fungal pathogens. Biofouling 2017, 33, 881–891. [Google Scholar] [CrossRef]
- Baindara, P.; Singh, N.; Ranjan, M.; Nallabelli, N.; Chaudhry, V.; Pathania, G.L.; Sharma, N.; Kumar, A.; Patil, P.B.; Korpole, S. Laterosporulin10: A novel defensin like class iid bacteriocin from brevibacillus sp. strain SKDU10 with inhibitory activity against microbial pathogens. Microbiology 2016, 162, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Accelrys Software Inc. Discovery Studio Modeling Environment, Release 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2009. [Google Scholar]
- Kar, S.; Drew, M.G.B.; Pramanik, A. Formation of Vesicles Through Solvent Assisted Self-Assembly of Hydrophobic Pentapeptides: Encapsulation and pH Responsive Release of Dyes by the Vesicles (Supplementary Material). Protein Pept. Lett. 2011, 18, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar]
- Luong, H.X.; Thanh, T.T.; Tran, T.H. Antimicrobial peptides—Advances in development of therapeutic applications. Life Sci. 2020, 260, 118407. [Google Scholar]
- Roy, A.; Franco, O.; Mandal, S. Biomedical Exploitation of Self Assembled Peptide Based Nanostructures. Curr. Protein Pept. Sci. 2013, 14, 580–587. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baindara, P.; Roy, D.; Mandal, S.M. CycP: A Novel Self-Assembled Vesicle-Forming Cyclic Antimicrobial Peptide to Control Drug-Resistant S. aureus. Bioengineering 2024, 11, 855. https://doi.org/10.3390/bioengineering11080855
Baindara P, Roy D, Mandal SM. CycP: A Novel Self-Assembled Vesicle-Forming Cyclic Antimicrobial Peptide to Control Drug-Resistant S. aureus. Bioengineering. 2024; 11(8):855. https://doi.org/10.3390/bioengineering11080855
Chicago/Turabian StyleBaindara, Piyush, Dinata Roy, and Santi M. Mandal. 2024. "CycP: A Novel Self-Assembled Vesicle-Forming Cyclic Antimicrobial Peptide to Control Drug-Resistant S. aureus" Bioengineering 11, no. 8: 855. https://doi.org/10.3390/bioengineering11080855
APA StyleBaindara, P., Roy, D., & Mandal, S. M. (2024). CycP: A Novel Self-Assembled Vesicle-Forming Cyclic Antimicrobial Peptide to Control Drug-Resistant S. aureus. Bioengineering, 11(8), 855. https://doi.org/10.3390/bioengineering11080855