
The Transformation Experiment of Frederick Griffith I: Its Narrowing and Potential for the Creation of Novel Microorganisms

Abstract
:1. Introduction: Artificial Creation of Novel Microorganisms by the Exchange of Genomes
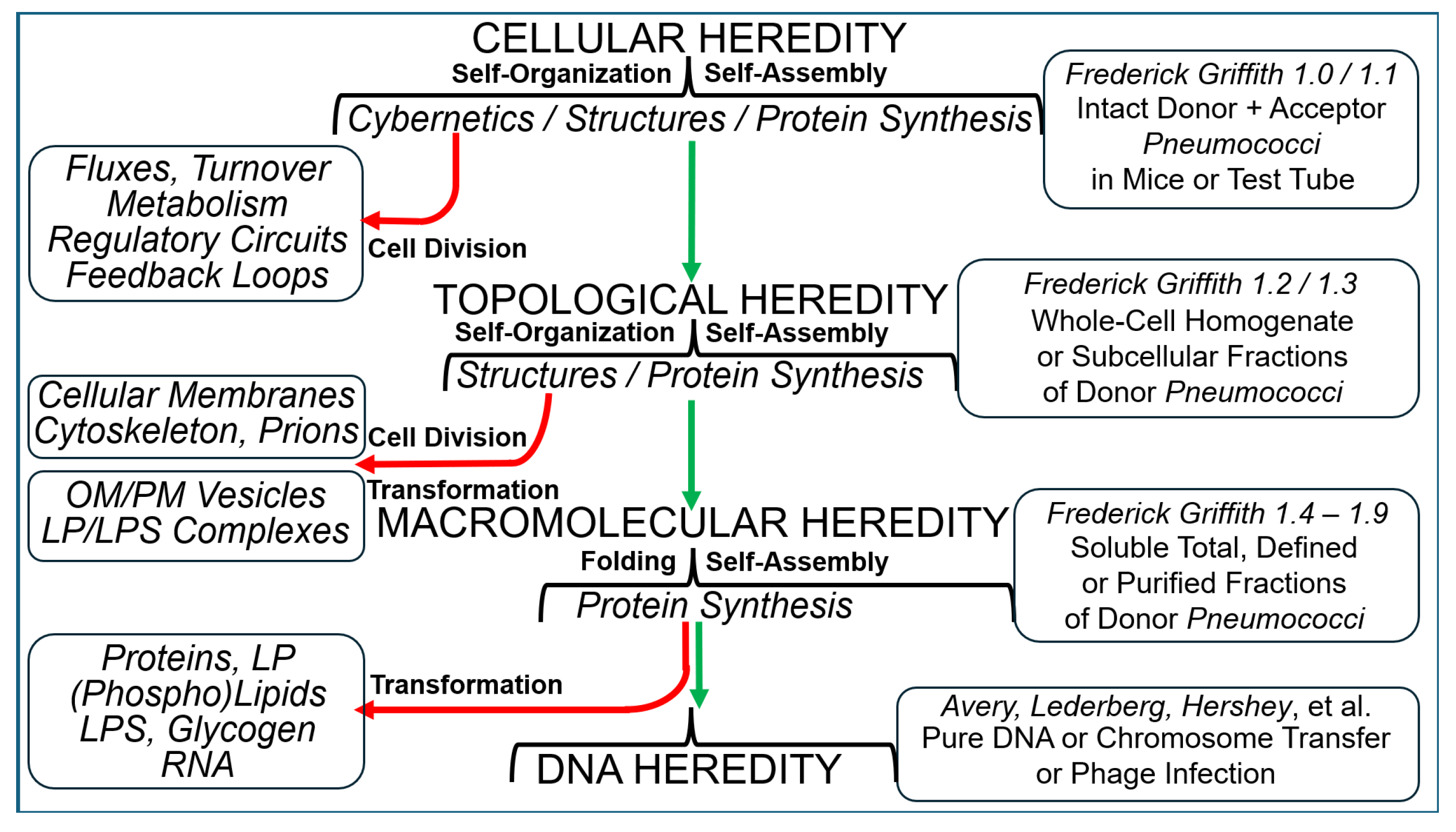
2. The Griffith Transformation Experiment (1.0) and Its Succeeding Designs
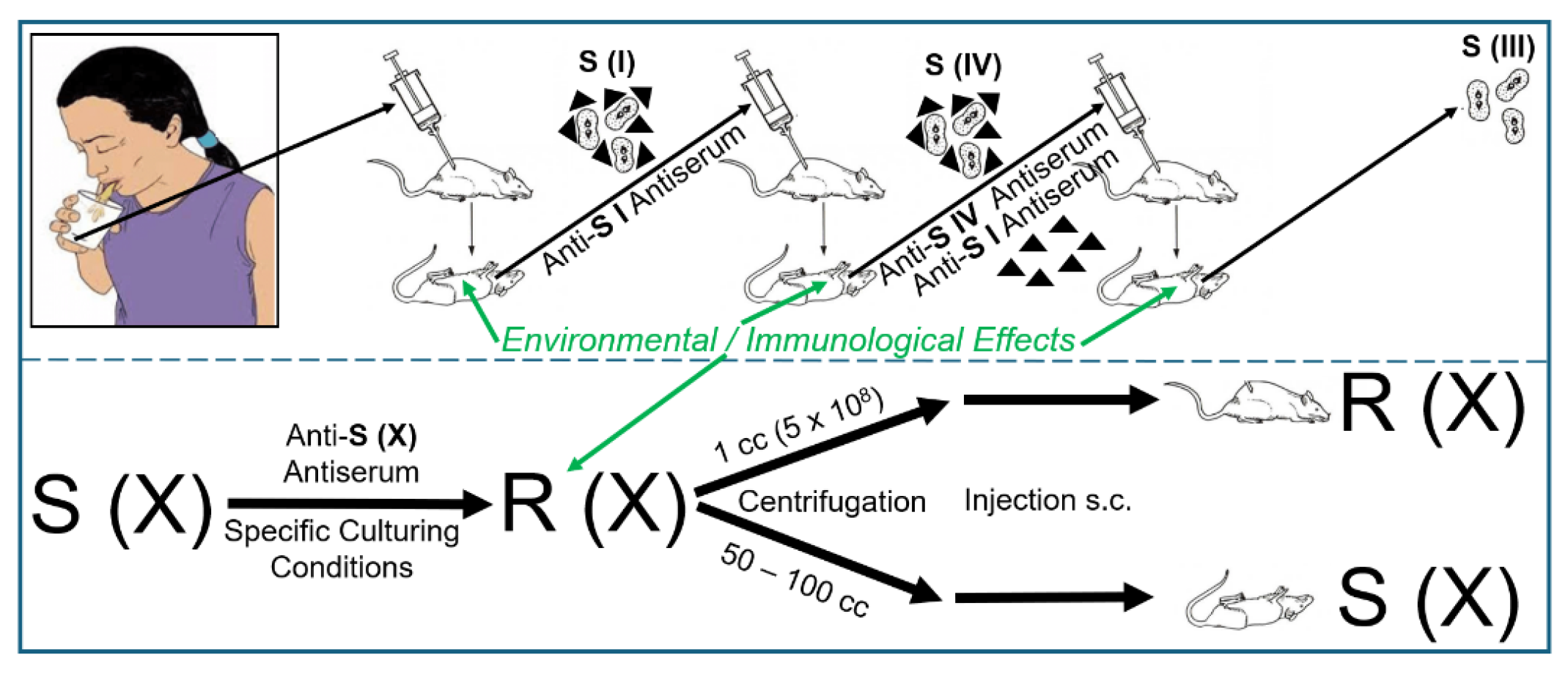
2.1. The Griffith Experiment in Mice
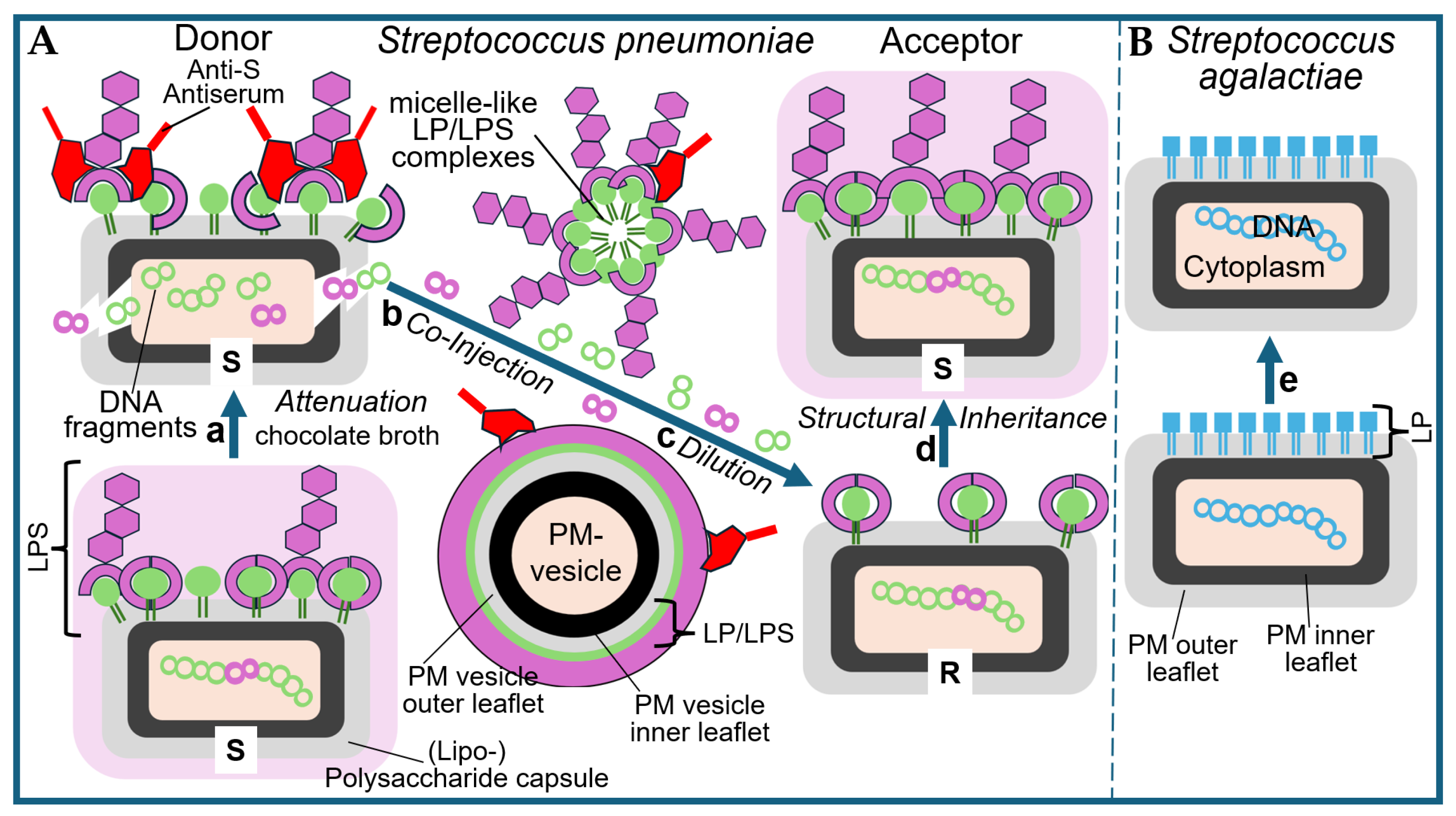
2.1.1. Bacterial Membrane Vesicles as a Putative Transforming Principle
2.1.2. Extracellular LP/LPS Complexes as a Putative Transforming Principle
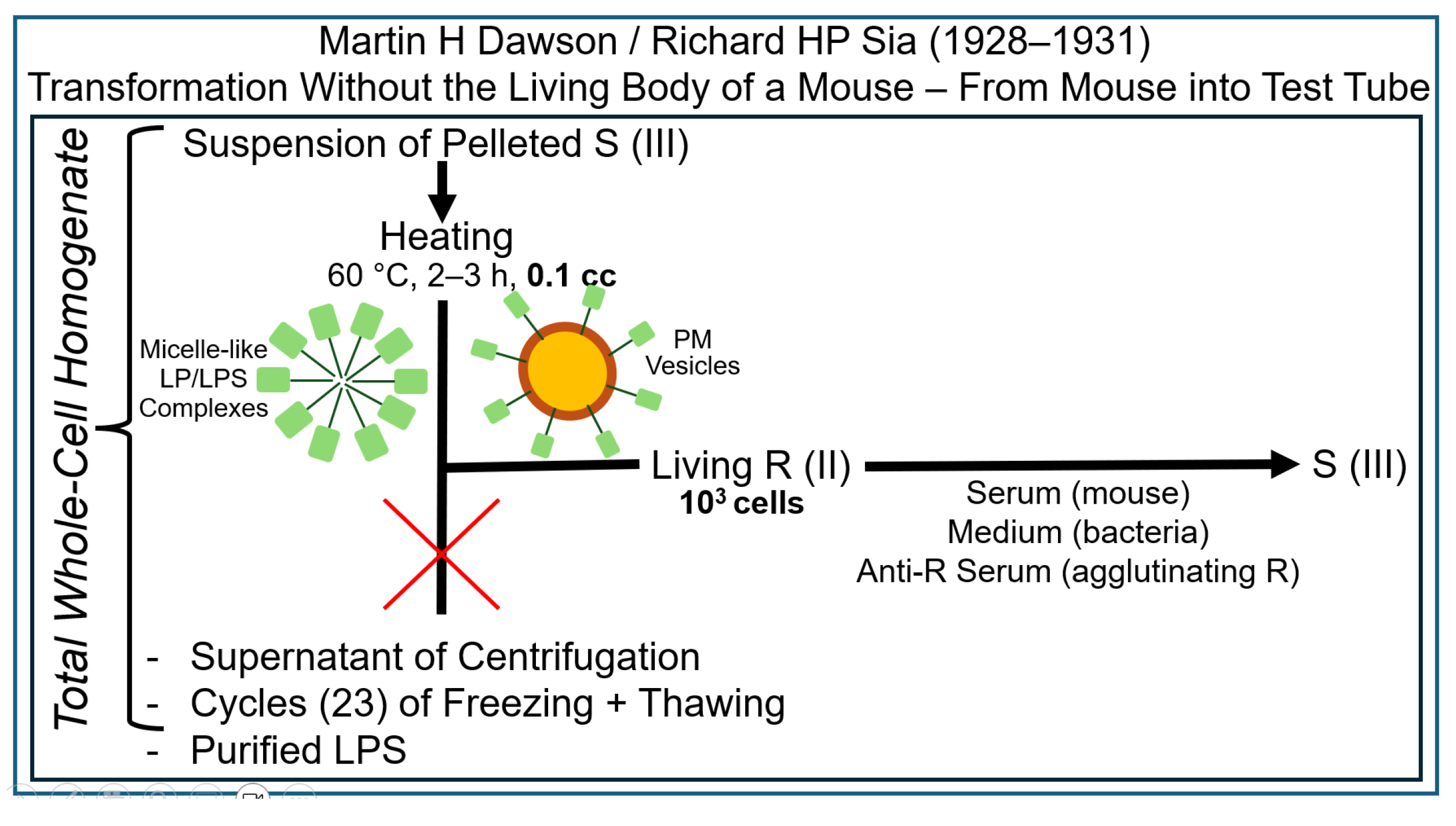
2.2. The Griffith Experiment in the Test Tube
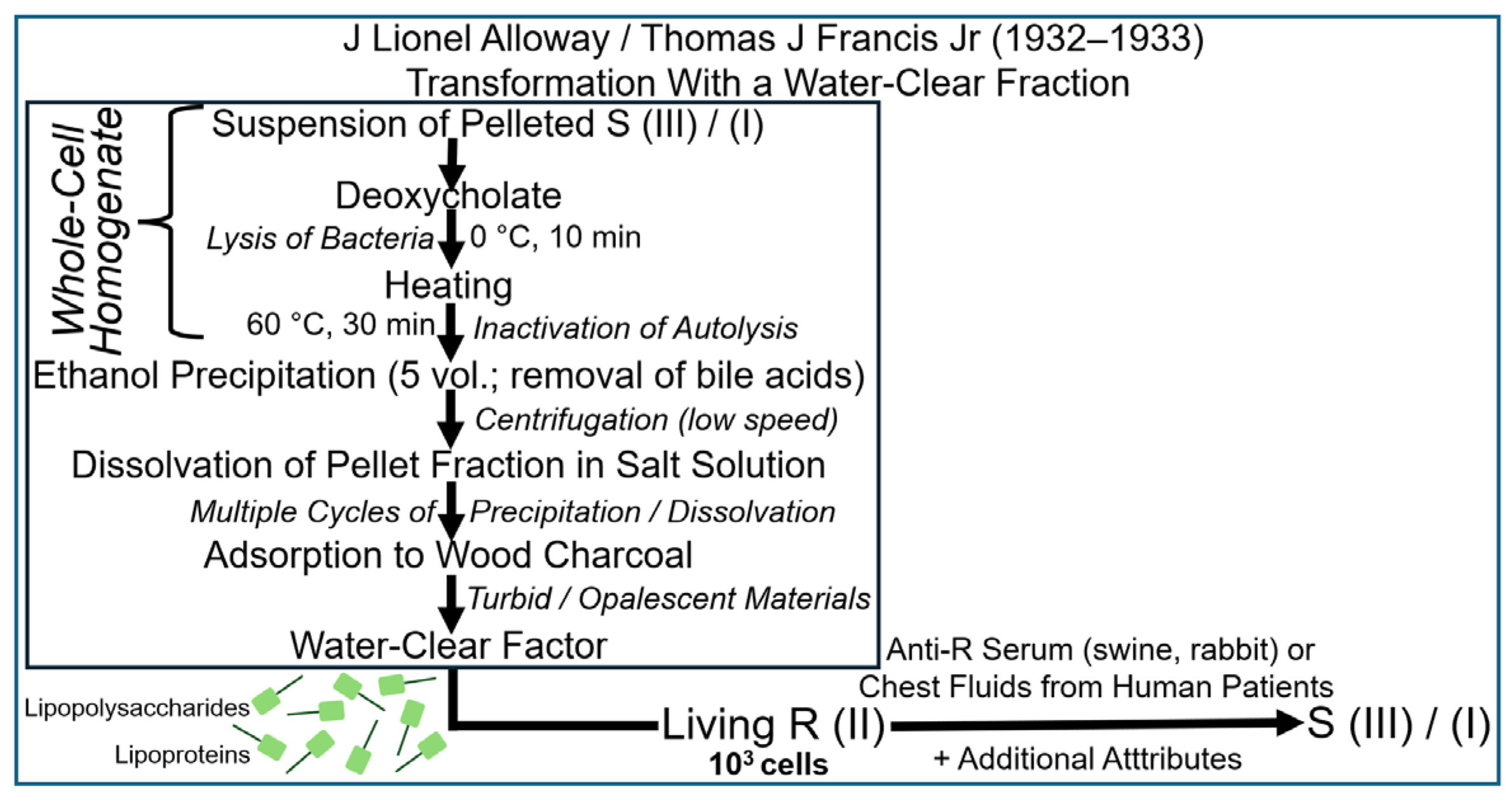
2.3. The Griffith Experiment with Subcellular Fractions
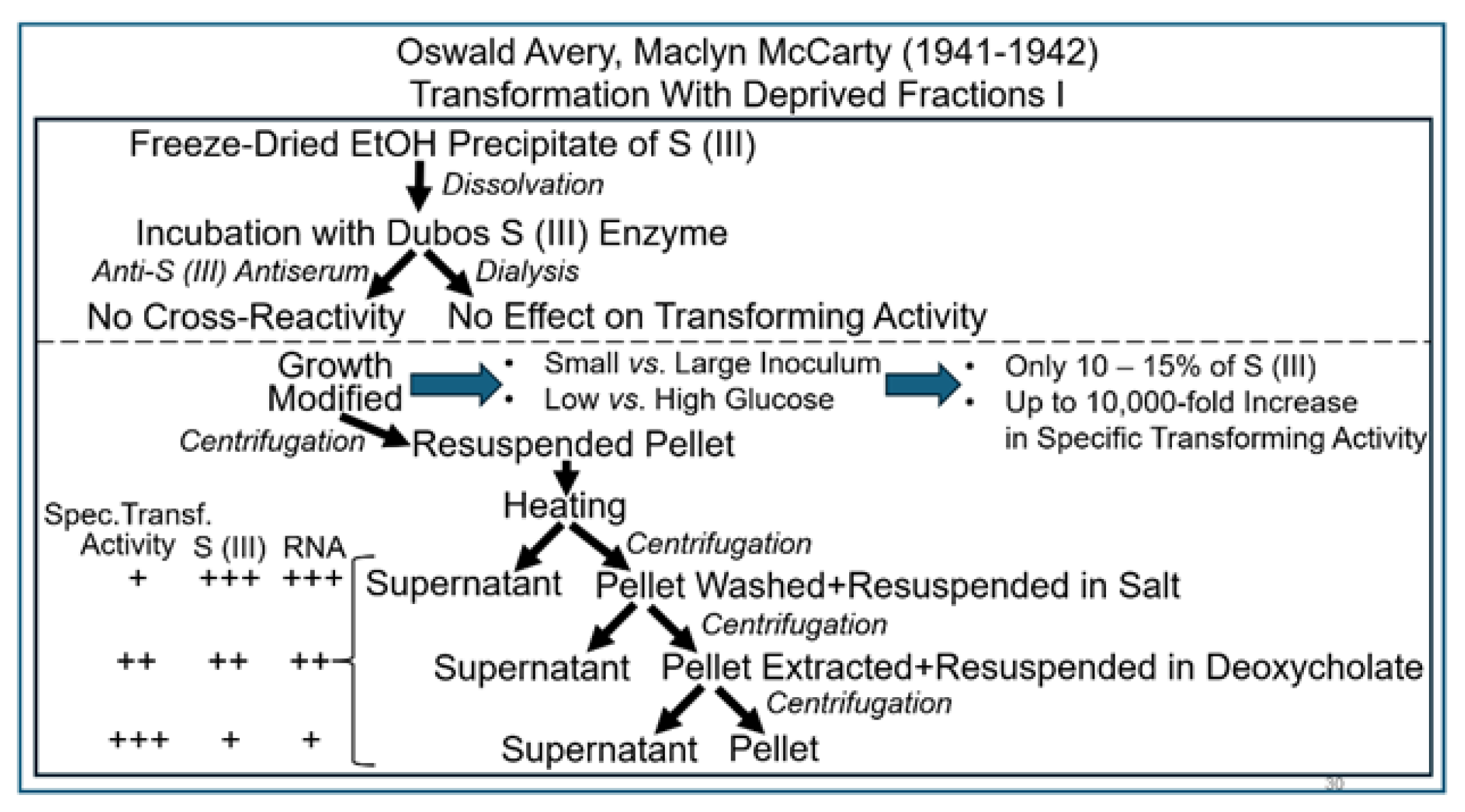
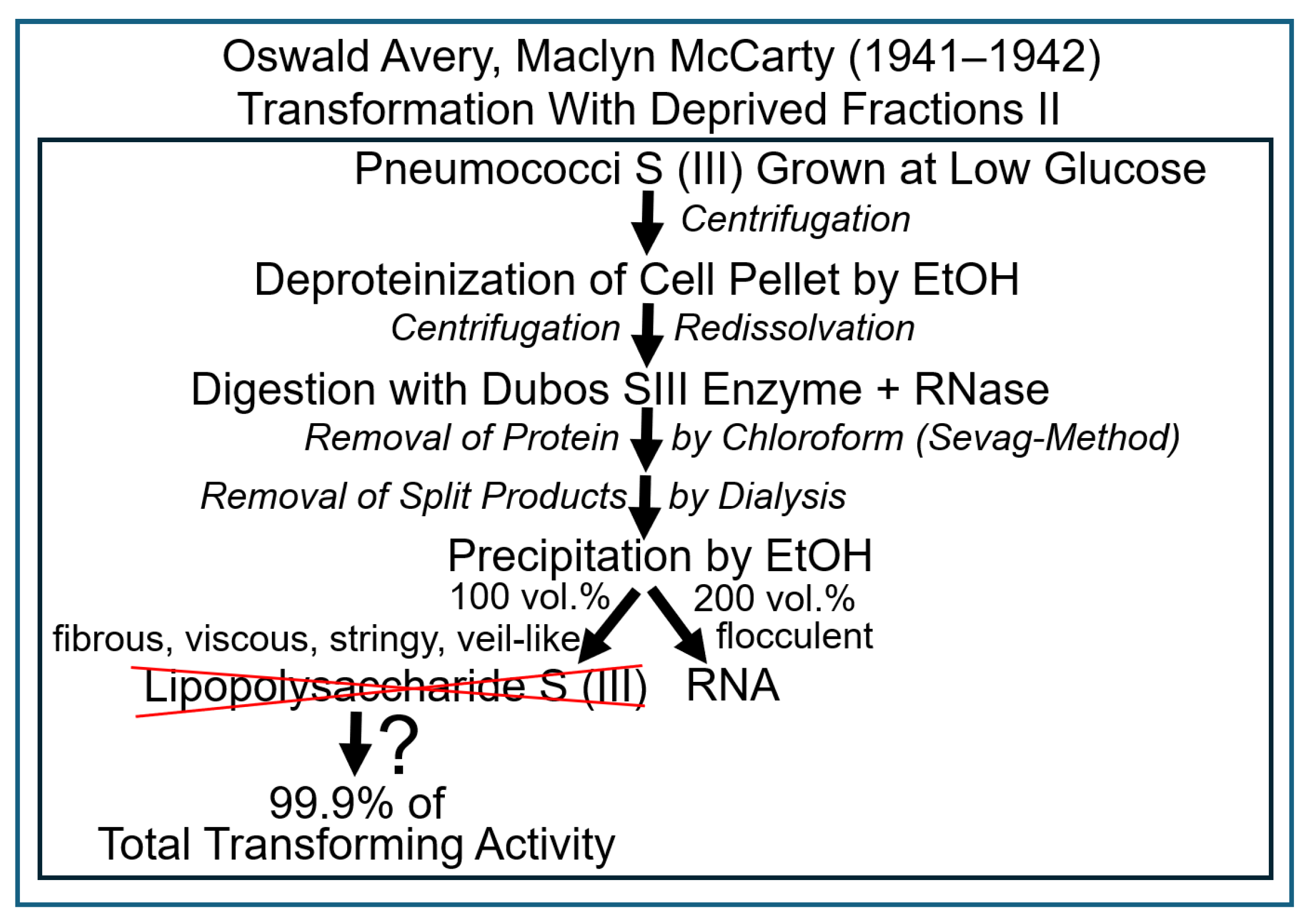
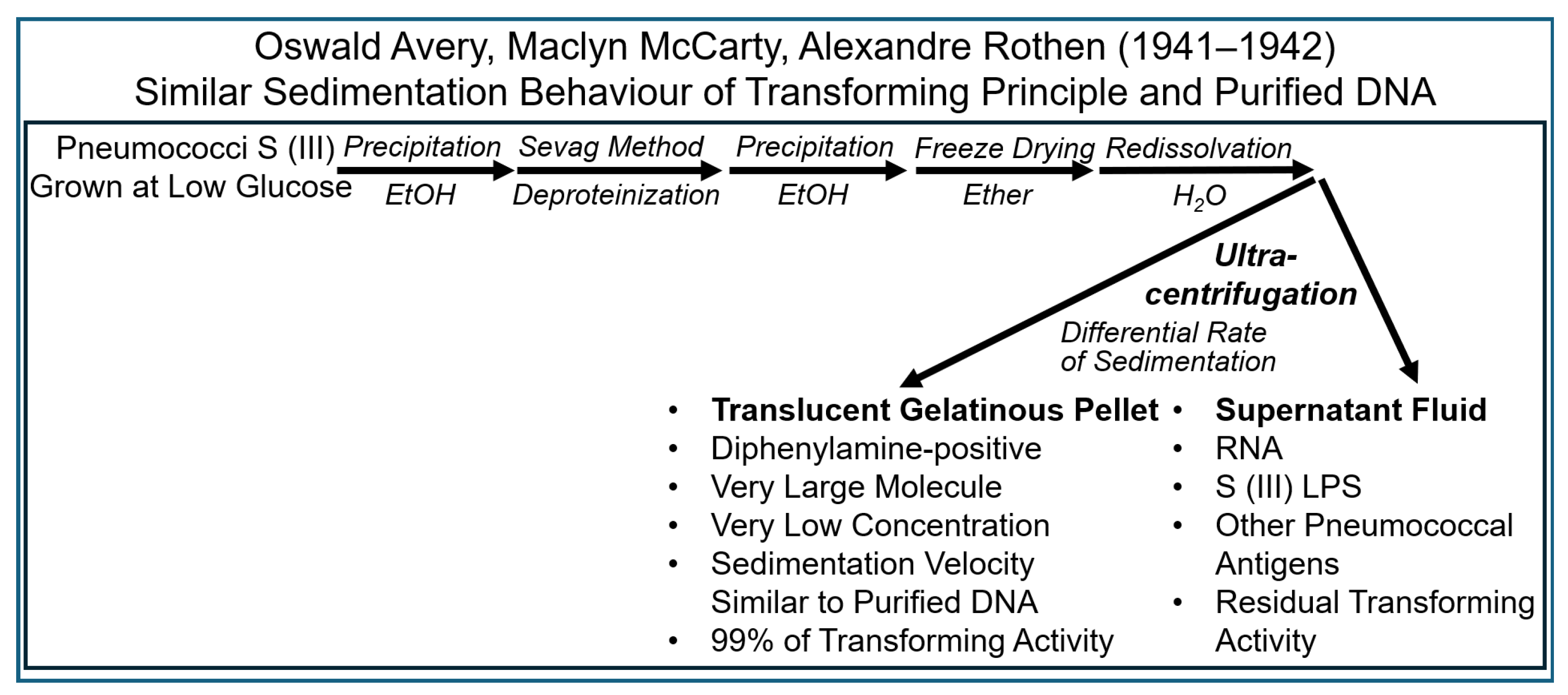
2.4. The Griffith Experiment with Macromolecules
2.5. The Griffith Experiment as the Foundation of Chromosome Transfer and Phage Infection
2.6. Transformation, Chromosome Transfer, Phage Infection and the Central Dogma of Molecular Biology
2.7. Bacterial Prions as a Putative Transforming Principle
3. Perspectives and Conclusions—Thought Styles About and Reception of Transformation
3.1. Conceivable Alternative Explanation of the Griffith Transformation Experiment
3.2. Putative (F)Actors That Affect the Identification of the Transforming Principle
3.3. Putative (F)Actors of Griffith’s Disinterest in Identification of the Transforming Principle
Funding
Conflicts of Interest
Abbreviations
| ANT | Actor-network theory |
| AR | Agential realism |
| LP | Lipoprotein(s) |
| LPS | Lipopolysaccharide(s) |
| OM | Outer membrane(s) |
| PM | Plasma membrane(s) |
| R/S | Rough/Smooth pneumococcal serotype |
| STS | Science and technology studies |
References
- Lartigue, C.; Glass, J., I; Alperovich, N.; Pieper, R.; Parmar, P.P.; Hutchison, C.A., 3rd; Smith, H.O.; Venter, J.C. Genome transplantation in bacteria: Changing one species to another. Science 2007, 317, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, C.; Vashee, S.; Algire, M.A.; Chuang, R.-Y.; Benders, G.A.; Ma, L.; Noskov, V.N.; Denisova, E.A.; Gibson, D.G.; Assad-Garcia, N.; et al. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 2009, 325, 1693–1696. [Google Scholar] [PubMed]
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.-Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [Google Scholar]
- Hutchison, C.A., 3rd; Chuang, R.-A.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal genome. Science 2016, 351, aad6253. [Google Scholar]
- Kannan, K.; Tsvetanova, B.; Chuang, R.-Y.; Noskov, V.N.; Assad-Garcia, N.; Ma, L.; Hutchison, C.A., 3rd; Smith, H.O.; Glass, J.I.; Merryman, C.; et al. One step engineering of the small-subunit ribosomal RNA using CRISPR/Cas9. Sci. Rep. 2016, 6, 30714. [Google Scholar]
- Zhang, L.-Y.; Chang, S.-H.; Wang, J. How to make a minimal genome for synthetic minimal cell. Protein Cell 2010, 1, 427–434. [Google Scholar]
- Glass, J.I.; Merryman, C.; Wise, K.S.; Hutchison, C.A., III; Smith, H.O. Minimal cells—Real and imaged. Cold Spring Harb. Perspect. Biol. 2017, 9, a023861. [Google Scholar] [CrossRef]
- Venter, J.C.; Glass, J.I.; Hutchinson, C.A., 3rd; Vashee, S. Synthetic chromosomes, genomes, viruses, and cells. Cell 2022, 185, 2708–2724. [Google Scholar] [CrossRef]
- Breuer, M.; Earnest, T.M.; Merryman, C.; Wise, K.S.; Sun, L.; Lynott, M.R.; Hutchison, C.A.; Smith, H.O.; Lapek, J.D.; Gonzalez, D.J.; et al. Essential metabolism for a minimal cell. Elife 2019, 8, e36842. [Google Scholar]
- Reyes-Prieto, M.; Gil, R.; Llabres, M.; Palmer-Rodriguez, P.; Moya, A. The metabolic building blocks of a minimal cell. Biology 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Schindler, D.; Walker, R.S.K.; Jiang, S.; Brooks, A.N.; Wang, Y.; Müller, C.A.; Cockram, C.; Luo, Y.; Garcia, A.; Schraivogel, D.; et al. Design, construction, and functional characterization of a tRNA neochromosome in yeast. Cell 2023, 186, 5237–5253. [Google Scholar] [CrossRef] [PubMed]
- Schindler, D.; Walker, R.S.K.; Cai, Y. Methodological advances enabled by the construction of a synthetic yeast genome. Cell Rep. Methods 2024, 4, 100761. [Google Scholar] [CrossRef] [PubMed]
- Moger-Reischer, R.Z.; Glass, J.I.; Wise, K.S.; Sun, L.; Bittencourt, D.M.C.; Lehmkuhl, B.K.; Schoolmaster, D.R., Jr.; Lych, M.; Lennon, J.T. Evolution of a minimal cell. Nature 2023, 620, 122–127. [Google Scholar] [CrossRef]
- Pelletier, J.F.; Glass, J.I.; Strychalski, E.A. Cellular mechanics during division of a genomically minimal cell. Trends Cell Biol. 2022, 32, 900–907. [Google Scholar] [CrossRef]
- Müller, G.A. The transformation experiment of Frederick Griffith: II. Inclusion of cellular heredity for the creation of novel microorganisms. Bioengineering 2025. to be submitted. [Google Scholar]
- Neufeld, F.; Levinthal, W. Beiträge zur Variabilität der Pneumokokken. Z. Immunitätforsch. 1928, 55, 324–340. [Google Scholar]
- Griffith, F. Types of haemolytic streptococci in relation to scarlet fever (second report). J. Hyg. 1927, 26, 363–373. [Google Scholar]
- Arkwright, J.A. Variation in bacteria in relation to agglutination both by salts and by specific serum. J. Pathol. Bacteriol. 1921, 24, 36–60. [Google Scholar]
- Lederberg, J. Bacterial variation since Pasteur. Rummaging in the Attic: Antiquarian ideas of transmissible heredity, 1880–1940. ASM News 1992, 58, 262–265. [Google Scholar]
- Griffith, F. The significance of pneumococcal types. J. Hyg. 1928, 27, 113–159. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; MacLeod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.; Lee, J.C. Staphylococcus aureus capsular polysaccharides. Clin. Microbiol. Rev. 2004, 17, 218–234. [Google Scholar] [CrossRef]
- Kohler, S.; Voß, F.; Gomez Mejia, A.; Brown, J.S.; Hammerschmidt, S. Pneumococcal lipoproteins in bacterial fitness, virulence, and immune evasion. FEBS J. 2016, 590, 3820–3839. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Simon, A.; Titball, R.W.; Michell, S.L. Lipoproteins of bacterial pathogens. Infect. Immun. 2011, 79, 548–561. [Google Scholar] [CrossRef]
- Briaud, P.; Carroll, R.K. Extracellular vesicle biogenesis and functions in gram-positive bacteria. Infect. Immun. 2020, 88, e00433-20. [Google Scholar] [CrossRef]
- Lee, J.; Kim, O.Y.; Gho, Y.S. Proteomic profiling of gram-negative bacterial outer membrane vesicles: Current perspectives. Proteomics 2016, 10, 897–909. [Google Scholar] [CrossRef]
- Toyofuku, M.; Nomura, N.; Eberl, I. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 2019, 17, 13–24. [Google Scholar] [CrossRef]
- Dineshkumar, K.; Aparna, V.; Wu, L.; Abdelaziz, M.H.; Su, Z.; Wang, S.; Xu, H. Bacterial bug-out bags: Outer membrane vesicles and their proteins and functions. J. Microbiol. 2020, 58, 531–542. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from gram-negative bacteria: Biogenesis and function. Nat. Rev. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, T.; Nomura, N.; Toyofuku, M. Cracking open bacterial membrane vesicles. Front. Microbiol. 2020, 10, 3026. [Google Scholar] [CrossRef] [PubMed]
- Sartorio, M.G.; Pardue, E.J.; Feldman, M.F.; Haurat, M.F. Bacterial outer membrane vesicles: From discovery to applications. Annu. Rev. Microbiol. 2021, 75, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Juodeikis, R.; Carding, S.R. Outer membrane vesicles: Biogenesis, function, and issues. Microbiol. Mol. Biol. Rev. 2022, 86, e00032-22. [Google Scholar] [CrossRef]
- Toyofuku, M.; Schild, S.; Kaparakis-Liaskos, M.; Eberl, L. Composition and functions of bacterial membrane vesicles. Nat. Rev. Microbiol. 2023, 21, 415–430. [Google Scholar] [CrossRef]
- Bishop, D.G.; Work, E. An extracellular glycolipid produced by Escherichia coli grown under lysine-limiting conditions. Biochem. J. 1965, 96, 567–576. [Google Scholar] [CrossRef]
- Haurat, M.F.; Elhenawy, W.; Feldman, M.F. Prokaryotic membrane vesicles: New insights on biogenesis and biological roles. Biol. Chem. 2015, 396, 95–109. [Google Scholar] [CrossRef]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef]
- Domingues, S.; Nielsen, K.M. Membranes vesicles and horizontal gene transfer. Curr. Opin. Microbiol. 2017, 38, 16–21. [Google Scholar] [CrossRef]
- Grüll, M.P.; Mulligan, M.E.; Lang, A.S. Small extracellular particles with big potential for horizontal gene transfer: Membrane vesicles and gene transfer agents. FEMS Microbiol. Lett. 2018, 365, fny192. [Google Scholar] [CrossRef]
- Tzipilevich, E.; Habuska, M.; Ben Yehuda, S. Aquisition of phage sensitivity by bacteria through exchange of phage receptors. Cell 2017, 168, 186–199.e12. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Herling, A.W.; Stemmer, K.; Lechner, K.; Lechner, A. Chip-based sensing for release of unprocessed cell surface proteins in vitro and in serum and its (patho)physiological relevance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E212–E233. [Google Scholar] [PubMed]
- Müller, G.A.; Ussar, S.; Tschöp, M.H.; Müller, T.D. Age-dependent membrane release and degradation of full-length glycosylphosphatidylinositol-anchored proteins in rats. Mech. Ageing Dev. 2020, 190, 111307. [Google Scholar]
- Müller, G.A. Glycosylphosphatidylinositol-Anchored Proteins and Their Release from Cells—From Phenomenon to Meaning; Nova Science Publishers—Biochemistry Research Trends: New York, NY, USA, 2018; pp. 104–115. [Google Scholar]
- Müller, G.A. The release of glycosylphosphatidylinositol-anchored proteins from the cell surface. Arch. Biochem. 2018, 656, 1–18. [Google Scholar]
- Müller, G.A.; Müller, T.D. (Patho)Physiology of glycosylphosphatidylinositol-anchored proteins I: Localization at plasma membranes and extracellular compartments. Biomolecules 2023, 13, 855. [Google Scholar] [CrossRef]
- Müller, G.A.; Müller, T.D. Transfer of membrane(s) matter(s)—Non-genetic inheritance of (metabolic) phenotypes? Front. Mol. Biosci. 2024, 11, 1347397. [Google Scholar] [CrossRef]
- Panakova, D.; Sprong, H.; Marois, E.; Thiele, C.; Eaton, S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005, 435, 58–65. [Google Scholar] [CrossRef]
- Neumann, S.; Harterink, M.; Sprong, H. Hitch-hiking between cells on lipoprotein particles. Traffic 2007, 8, 331–338. [Google Scholar] [CrossRef]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef]
- Müller, G.A.; Müller, T.D. Biological role of the intercellular transfer of glycosylphosphatidylinositol-anchored proteins: Stimulation of lipid and glycogen synthesis. Int. J. Mol. Sci. 2023, 23, 7418. [Google Scholar]
- Müller, G.A.; Müller, T.D. Transfer of proteins from cultured human adipose to blood cells and induction of anabolic phenotype are controlled by serum, insulin and sulfonylurea drugs. Int. J. Mol. Sci. 2023, 24, 4825. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Müller, T.D. A “poly-matter network” conception of biological inheritance. Genetica 2024, 152, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. Glycosylphosphatidylinositol-anchored proteins as non-DNA matter of inheritance: From molecular to cell to philosophical biology. Acad. Mol. Biol. Genom. 2024, 1, 1–36. [Google Scholar] [CrossRef]
- Dochez, A.R.; Avery, O.T. The elaboration of a specific soluble substance by pneumococcus during growth. J. Exp. Med. 1917, 26, 477–493. [Google Scholar]
- Heidelberger, M.; Goebel, W.F. The soluble specific substance of pneumococcus. IV. On the nature of the specific polysaccharide of Type III pneumococcus. J. Biol. Chem. 1926, 70, 613–624. [Google Scholar]
- Goebel, W.F. Studies on antibacterial immunity induced by artificial antigens. I. Immunity to experimental pneumococcal infection with an antigen containing cellobiuronic acid. J. Exp. Med. 1939, 69, 353–364. [Google Scholar]
- Geno, K.A.; Gilbert, G.L.; Song, J.Y.; Skovsted, I.C.; Klugman, K.P.; Jones, C.; Konradsen, H.B.; Nahm, M.H. Pneumococcal capsules and their types: Past, present, and future. Clin. Microbiol. Rev. 2015, 28, 871–898. [Google Scholar]
- Calix, J.J.; Brady, A.M.; Du, V.Y.; Saad, J.S.; Nahm, M.H. Spectrum of pneumococcal serotype 11A variants results from incomplete loss of capsule O-acetylation. J. Clin. Microbiol. 2014, 52, 758–765. [Google Scholar] [CrossRef]
- Rajagopal, M.; Walker, S. Envelope structures of gram-positive bacteria. Curr. Top. Microbiol. Immunol. 2017, 404, 1–44. [Google Scholar] [CrossRef]
- Vollmer, W.; Massidda, O.; Tomasz, A. The cell wall of Streptococcus pneumoniae. Microbiol. Spectr. 2019, 7, 284–303. [Google Scholar] [CrossRef]
- Pollock, M.R. The changing concept of organism in microbiology. Prog. Biophys. Mol. Biol. 1969, 19, 271–305. [Google Scholar] [PubMed]
- Olby, R. The Path to the Double Helix. The Discovery of DNA; Dover Publication: New York, NY, USA, 1994. [Google Scholar]
- Amsterdamska, O. Medical and biological constraints: Early research on variation in bacteriology. Soc. Stud. Sci. 1987, 17, 657–687. [Google Scholar] [PubMed]
- Eichmann, K.; Krause, R.M. Fred Neufeld and pneumococcal serotypes: Foundations for the discovery of the transforming principle. Cell. Mol. Life Sci. 2013, 70, 2225–2236. [Google Scholar] [PubMed]
- Rheinberger, H.-J. Epistemologie des Konkreten. Studien zur Geschichte der Modernen Biologie; Suhrkamp: Frankfurt am Main, Germany, 2006. [Google Scholar]
- Rheinberger, H.-J. Iterationen; Merve: Berlin, Germany, 2005. [Google Scholar]
- Hacking, I. Representing and Intervening. Introductory Topics in the Philosophy of Natural Science; Cambridge University Press: Cambridge, MA, USA; New York, NY, USA; Oakleigh, PA, USA, 1983. [Google Scholar]
- Methot, P.-O. Bacterial transformation and the origins of epidemics in the interwar period: The epidemiological significance of Fred Griffith’s “Transformation Experiment”. J. Hist. Biol. 2016, 49, 311–358. [Google Scholar]
- Dawson, M.H. The interconvertibility of “R” and “S” forms of pneumococcus. J. Exp. Med. 1928, 47, 577–591. [Google Scholar]
- Dawson, M.H. The transformation of pneumococcal types. I. The conversion of R forms of pneumococcus into S forms of the homologous type. J. Exp. Med. 1929, 50, 99–122. [Google Scholar]
- Dawson, M.H. The transformation of pneumococcus types. II. The interconvertibility of type-specific S pneumococci. J. Exp. Med. 1930, 51, 123–147. [Google Scholar]
- Dawson, M.H.; Sia, R.H.P. In vitro transformations of pneumococcal types. I. A technique for inducing transformations of pneumococcal types. I. A technique for inducing transformations of pneumococcal types in vitro. J. Exp. Med. 1931, 53, 681–699. [Google Scholar] [CrossRef]
- Sia, R.H.P.; Dawson, M.H. In vitro transformations of pneumococcal types. II. The nature of the factor responsible for the transformation of pneumococcal types. J. Exp. Med. 1931, 54, 701–710. [Google Scholar]
- Alloway, J.L. The transformation in vitro of R pneumococci into S forms of different specific types by the use of filtered pneumococcus extracts. J. Exp. Med. 1932, 55, 91–99. [Google Scholar]
- Alloway, J.L. Further observations on the use of pneumococcus extracts in effecting transformation of type in vitro. J. Exp. Med. 1933, 57, 265–278. [Google Scholar] [PubMed]
- MacLeod, C.M.; McCarty, M. The relation of a somatic factor to virulence of pneumococci. J. Clin. Investig. 1942, 21, 647. [Google Scholar]
- Sevag, M.G. Eine neue physikalische Enteiweissungsmethode zur Darstellung biologisch wirksamer Substanzen. Isolierung von Kohlenhydraten aus Hühnereiweiss und Pneumokokken. Biochem. Z. 1934, 273, 419. [Google Scholar]
- McCarty, M. Reminiscences of the early days of transformation. Annu. Rev. Genet. 1980, 14, 1–15. [Google Scholar]
- McCarty, M. The Transforming Principle—Discovering That Genes Are Made of DNA; Norton & Company: New York, NY, USA; London, UK, 1985. [Google Scholar]
- Avery, O.T.; Dubos, R. The protective action of a specific enzyme against type III pneumococcus infection in mice. J. Exp. Med. 1931, 54, 73–89. [Google Scholar]
- Mirsky, A.E.; Pollister, A.W. Chromosin, a desoxyribose nucleoprotein complex of the cell nucleus. J. Gen. Physiol. 1946, 30, 130–139. [Google Scholar]
- Caspersson, T.; Hammarsten, E.; Hammarsten, H. Interaction of proteins and nucleic acids. Transact. Faraday Soc. 1935, 31, 367–389. [Google Scholar] [CrossRef]
- Schultz, J. The evidence of the nucleoprotein nature of the gene. Cold Spring Harb. Symp. Quant. Biol. 1941, 9, 56–65. [Google Scholar] [CrossRef]
- Fischer, F.G.; Böttger, I.; Lehmann-Echternacht, H. Über die Thymo-polynucleotidase aus Pancreas. Nucleinsäure. V. Z. Physiol. Chem. 1941, 271, 246–264. [Google Scholar] [CrossRef]
- Mirsky, A.E. Chromosomes and nucleoproteins. Adv. Enzymol. 1943, 3, 1–23. [Google Scholar]
- Caspersson, T. Methods for the determination of the absorption spectra of cell structures. J. R. Microscop. Soc. 1940, 60, 8–19. [Google Scholar]
- Casperson, T.; Schultz, J. Ribonucleic acids in both nucleus and cytoplasm, and the function of the nucleolus. Proc. Natl. Acad. Sci. USA 1940, 26, 507–515. [Google Scholar]
- Darlington, C.D. Chromosome chemistry and gene action. Nature 1942, 149, 66–71. [Google Scholar]
- Astbury, W.T. Protein and Virus Studies in Relation to the Problem of the Gene. In Proceedings of the 7th International Genetical Congress, Edinburgh, UK, 23–30 August 1939; Cambridge University Press: Cambridge, UK, 1941; p. 49. [Google Scholar]
- McCarty, M. Reversible inactivation of the substance inducing transformation of pneumococcal types. J. Exp. Med. 1945, 81, 501–514. [Google Scholar]
- McCarty, M. Purification and properties of desoxyribonuclease isolated from beef pancreas. J. Gen. Physiol. 1946, 29, 123–139. [Google Scholar]
- McCarty, M.; Avery, O.T. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. II. Effect of desoxyribonuclease on the biological activity of the transforming substance. J. Exp. Med. 1946, 83, 89–96. [Google Scholar]
- McCarty, M.; Avery, O.T. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. III. An improved method for the isolation of the transforming substance and its application to pneumococcus types II, III, and VI. J. Exp. Med. 1946, 83, 97–104. [Google Scholar]
- McCarty, M. Chemical nature and biological specificity of the substance inducing transformation of pneumococcal types. Bacteriol. Rev. 1946, 10, 63–71. [Google Scholar]
- McCarty, M.; Taylor, H.E.; Avery, O.T. Biochemical studies of environmental factors essential in transformation of pneumococcal types. Cold Spring Harb. Lab. Quant. Biol. 1946, 11, 177–183. [Google Scholar]
- Taylor, H.E. Additive effects of certain transforming agents from some variants of pneumococcus. J. Exp. Med. 1949, 89, 399–424. [Google Scholar] [CrossRef]
- Austrian, R.; MacLeod, C.M. Acquisition of M protein by pneumococci through transformation reactions. J. Exp. Med. 1949, 89, 451–460. [Google Scholar] [PubMed]
- Astbury, W.T. X-ray studies of nucleic acids. Symp. Soc. Exp. Biol. 1947, 1, 66–71. [Google Scholar]
- Caspersson, T. The relations between nucleic acid and protein synthesis. Symp. Soc. Exp. Biol. 1947, 1, 127–132. [Google Scholar]
- Gulland, J.M. Structures of nucleic acids. Symp. Soc. Exp. Biol. 1947, 1, 1–9. [Google Scholar]
- Stacey, M. Bacterial nucleic acids and nucleoproteins. Symp. Soc. Exp. Biol. 1947, 1, 86–93. [Google Scholar]
- Brachet, J. La detection histochimique des acides pentose-nucleiques. C. R. Soc. Biol. 1940, 133, 80–87. [Google Scholar]
- Brachet, J. Nucleic acids in the cell and the embryo. Symp. Soc. Exp. Biol. 1947, 1, 207–212. [Google Scholar]
- Chargaff, E. Chemical specificity of nucleic acids and mechanism of their enzymatic degradation. Experientia 1950, 6, 201–207. [Google Scholar]
- Vischer, E.; Zamenhof, S.; Chargaff, E. Microbial nucleic acids: The desoxypentose nucleic acids of avian tubercle bacilli and yeast. J. Biol. Chem. 1949, 177, 429–435. [Google Scholar]
- Zamenhof, S.; Brewerman, G.; Chargaff, E. On the desoxypentose nucleic acids from several micro-organisms. Biochim. Biophys. Acta 1952, 9, 402–409. [Google Scholar]
- Chargaff, E.; Lipshitz, R.; Green, C.; Hodes, M.E. The composition of desoxyribonucleic acid of salmon sperm. J. Biol. Chem. 1951, 192, 223–230. [Google Scholar] [PubMed]
- Daly, M.M.; Alfrey, V.G.; Mirsky, A.E. Purine and pyrimidine contents of some desoxypentose nucleic acids. J. Gen. Physiol. 1950, 33, 497–503. [Google Scholar] [PubMed]
- Wyatt, G.R. The nucleic acids of some insect viruses. J. Gen. Physiol. 1952, 36, 201–206. [Google Scholar] [PubMed]
- Hotchkiss, R.D. Etudes chimiques sur le facteur transformant du pneumocoque. Colloq. Int. Cent. Natl. Rech. Sci. 1949, 8, 57–64. [Google Scholar]
- Tatum, E.L.; Lederberg, J. Gene recombination in the bacterium Escherichia coli. J. Bact. 1947, 53, 673–678. [Google Scholar] [CrossRef]
- Lederberg, J.; Lederberg, E.M.; Zinder, N.D.; Lively, E.R. Recombination analysis of bacterial heredity. Cold Spring Harb. Symp. Quant. Biol. 1951, 16, 413–421. [Google Scholar] [CrossRef]
- Zinder, N.D.; Lederberg, J. Genetic exchange in Salmonella. J. Bact. 1952, 64, 679–685. [Google Scholar] [CrossRef]
- Kaudewitz, F. Molekular- und Mikroben-Genetik; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1973. [Google Scholar]
- Anderson, T.F.; Wollman, E.L.; Jacob, F. Sur les processus de conjugaison et de recombination génétique chez E. coli. III. Aspects morphologiques en microscopie électronique. Ann. Inst. Pasteur 1957, 93, 450–458. [Google Scholar] [PubMed]
- Watanabe, T. Infectious drug resistance. Sci. Am. 1967, 217, 19–26. [Google Scholar] [CrossRef]
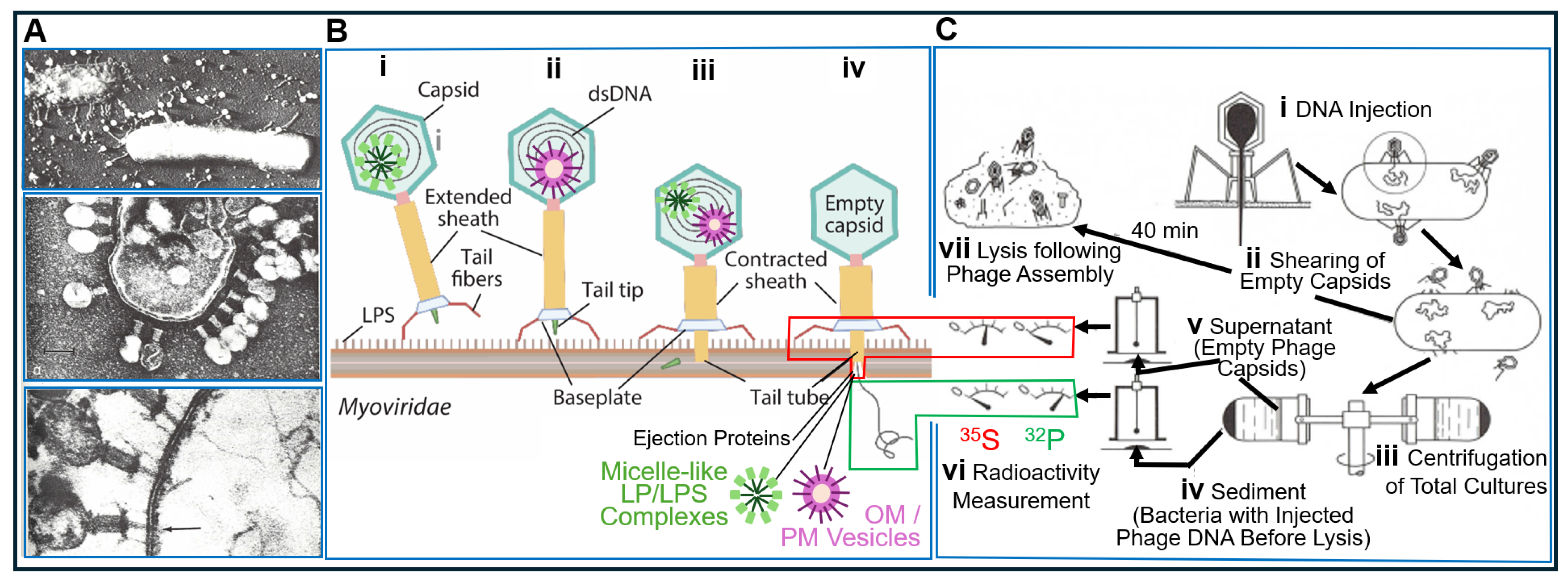
- Hershey, A.D.; Chase, M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar]
- Simon, I.D.; Anderson, T.F. The infection of Escherichia coli by T2 and T4 bacteriophages as seen in the electron microscope. I. Attachment and penetration. Virology 1967, 32, 279–297. [Google Scholar] [PubMed]
- Molineux, I.J. Fifty-three years since Hershey and Chase; much ado about pressure but which pressure is it? Virology 2006, 344, 221–229. [Google Scholar] [PubMed]
- Iglesias, S.M.; Li, F.; Briani, F.; Cingolani, G. Viral genome delivery across bacterial cell surfaces. Annu. Rev. Microbiol. 2024, 78, 125–145. [Google Scholar] [PubMed]
- Dunne, M.; Hupfeld, M.; Klumpp, J.; Loessner, M.J. Molecular basis of bacterial host interactions by Gram-positive targeting bacteriophages. Viruses 2018, 10, 397. [Google Scholar] [CrossRef]
- Guasch, A.; Pous, J.; Ibarra, B.; Gomis-Ruth, F.X.; Valpuesta, J.M.; Sousa, N.; Carrascosa, J.L.; Coll, M. Detailed architecture of a DNA translocating machine: The high resolution structure of the bacteriophage o29 connector particle. J. Mol. Biol. 2002, 315, 663–676. [Google Scholar]
- Guilhard, G.; Boulanger, P.; Letellier, I. Involvement of phage T5 tail proteins and contact sites between the outer and inner membrane of Escherichia coli in phage T5 DNA injection. J. Biol. Chem. 1992, 267, 3173–3178. [Google Scholar]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossman, M.G. Structure of the cell-puncturing device of bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar]
- Kemp, P.; Gupta, M.; Molineux, I.J. Bacteriophage T7 DNA ejection into cells is initiated by an enzyme-like mechanism. Mol. Microbiol. 2004, 53, 1251–1265. [Google Scholar]
- Kemp, P.; Garcia, I.R.; Molineux, I.J. Changes in bacteriophage T7 virion structure at the initiation of infection. Virology 2005, 340, 307–317. [Google Scholar]
- Leiman, P.G.; Chipman, P.R.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell 2004, 118, 419–429. [Google Scholar]
- Purohit, P.K.; Inamdar, M.M.; Grayson, P.D.; Squires, T.M.; Kondev, J.; Phillips, R. Forces during bacteriophage DNA packaging and ejection. Biophys. J. 2005, 88, 851–856. [Google Scholar] [PubMed]
- Roesner, C.A.; Ihler, G.M. Formation of transmembrane channels in liposomes during injection of lambda DNA. J. Biol. Chem. 1986, 261, 386–390. [Google Scholar]
- Kutter, E.; Bryan, D.; Ray, G.; Brewster, E.; Blasdel, B.; Guttman, B. From host to phage metabolism: Hot tales of phage T4’s takeover of E. coli. Viruses 2018, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Mangenot, S.; Hochrein, M.; Radler, J.; Letellier, I. Real-time imaging of DNA ejection from single phage particles. Curr. Biol. 2005, 15, 430–435. [Google Scholar]
- Kushner, D.J. Self-assembly of biological structures. Bacteriol. Rev. 1969, 33, 302–345. [Google Scholar]
- Amos, L.A.; Finch, J.T. Aaron Klug and the revolution in biomolecular structure determination. Trends Cell Biol. 2004, 14, 148–152. [Google Scholar]
- Schuster, T.M.; Scheele, R.B.; Adams, M.L.; Shire, S.J.; Steckert, J.J.; Potschka, M. Studies on the mechanism of assembly of tobacco mosaic virus. Biophys. J. 1980, 32, 313–329. [Google Scholar]
- De Gier, J.W.; Valent, Q.A.; von Heinje, G.; Luirink, J. The E. coli SRP: Preferences of a targeting factor. FEBS Lett. 1997, 408, 1–4. [Google Scholar]
- Wild, K.; Weichenrieder, O.; Strub, K.; Sinning, I.; Cusack, S. Towards the structure of the mammalian signal recognition particle. Curr. Opin. Struct. Biol. 2002, 12, 72–81. [Google Scholar]
- Leung, E.; Brown, J.D. Biogenesis of the signal recognition particle. Biochem. Soc. Trans. 2010, 38, 1093–1098. [Google Scholar]
- Massenet, S. In vivo assembly of eukaryotic signal recognition particle: A still enigmatic process involving the SMN complex. Biochimie 2019, 164, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Varela, F. Principles of Biological Autonomy; Appleton & Lange: New York, NY, USA, 1979. [Google Scholar]
- Kirschner, M.W.; Gerhart, J.C. The Plausibility of Life—Resolving Darwin’s Dilemma. Yale University Press: New Haven, CT, USA; London, UK, 2005. [Google Scholar]
- Kirschner, M.; Mitchison, T. Beyond self-assembly: From microtubules to morphogenesis. Cell 1986, 45, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Malinovska, L.; Kroschwald, S.; Alberti, S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim. Biophys. Acta 2013, 1834, 918–931. [Google Scholar] [CrossRef]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation prone” and “aggregation susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Wang, F.; Ma, J. Role of lipid in forming an infectious prion? Acta Biochim. Biophys. Sin. 2013, 45, 485–493. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 2005, 6, 435–450. [Google Scholar] [CrossRef]
- Jager, K.; Orozco-Hidalgo, M.T.; Springstein, B.L.; Joly-Smith, E.; Papazotos, F.; McDonough, E.K.; Fleming, E.; McCallum, G.; Yuan, A.H.; Hilfinger, A.; et al. Measuring prion propagation in single bacteria elucidates a mechanism of loss. Proc. Natl. Acad. Sci. USA 2023, 120, e2221539120. [Google Scholar] [CrossRef]
- Staniforth, G.L.; Tuite, M.F. Fungal prions. Prog. Mol. Biol. Transl. Sci. 2012, 107, 417–456. [Google Scholar]
- Toombs, J.A.; McCarty, B.R.; Ross, E.D. Compositional determinants of prion formation in yeast. Mol. Cell Biol. 2010, 30, 319–332. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Coquel, A.-S.; Moreno-del Alamo, M.; Chen, P.; Song, X.; Serrano, A.M.; Fernandez-Tresguerres, M.E.; Moreno-Diaz de la Espina, S.; Lindner, A.B.; Giraldo, R. Direct assessment in bacteria of prionoid propagation and phenotype selection by HSP70 chaperone. Mol. Microbiol. 2014, 91, 1070–1987. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.H.; Garrity, S.J.; Nako, E.; Hochschild, A. Prion propagation can occur in a prokaryote and requires the ClpB chaperone. eLife 2014, 3, e02949. [Google Scholar] [CrossRef]
- Iglesias, V.; de Groot, N.S.; Ventura, S. Computational analysis of candidate prion-like proteins in bacteria and their role. Front. Microbiol. 2015, 6, 1123. [Google Scholar] [CrossRef] [PubMed]
- Fleming, E.; Yuan, A.H.; Heller, D.M.; Hochschild, A. A bacteria-based genetic assay detects prion formation. Proc. Natl. Acad. Sci. USA 2019, 116, 4605–4610. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Taylor, J.D.; Matthews, S.J. New insight into the molecular control of bacterial functional amyloids. Front. Cell. Infect. Microbiol. 2015, 5, 33. [Google Scholar] [CrossRef]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef]
- Graether, S.P.; Slupsky, C.M.; Sykes, B.D. Freezing of a fish antifreeze protein results in amyloid fibril formation. Biophys. J. 2003, 84, 552–557. [Google Scholar] [CrossRef]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef]
- Fernandez-Tresguerres, M.E.; Moreno-Diaz de la Espina, S.; Gasset-Rosa, F.; Giraldo, R. A DNA-promoted amyloid proteinopathy in Escherichia coli. Mol. Microbiol. 2010, 77, 1456–1469. [Google Scholar] [CrossRef]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aß) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [PubMed]
- Halfmann, R.; Alberti, S.; Krishnan, R.; Lyle, N.; O’Donnell, C.W.; King, O.D.; Berger, B.; Pappu, R.V.; Lindquist, S. Opposing effect of glutamine and asparagine govern prion formation by intrinsically disordered protein. Mol. Cell 2011, 43, 72–84. [Google Scholar] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [PubMed]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef]
- Harrison, P.M.; Gerstein, M. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol. 2003, 4, R40. [Google Scholar]
- Chapman, M.R.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef]
- Wang, X.; Chapman, M.R. Curli provide the template for understanding controlled amyloid propagation. Prion 2008, 2, 57–60. [Google Scholar]
- Schwartz, K.; Boles, B.R. Microbial amyloids—Functions and interactions within the host. Curr. Opin. Microbiol. 2013, 16, 93–99. [Google Scholar] [CrossRef]
- Hill, J.M.; Lukiw, W.J. Microbial-generated amyloids and Alzheimer’s disease (AD). Front. Aging Neurosci. 2015, 7, 9. [Google Scholar]
- Giraldo, R. The emergence of bacterial prions. PLoS Pathog. 2024, 20, e1012253. [Google Scholar] [CrossRef] [PubMed]
- Fink, G.R. A transforming principle. Cell 2005, 120, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Stanford, P.K. Darwin’s pangenesis and the problem of unconceived alternatives. Br. J. Philos. Sci. 2006, 57, 121–144. [Google Scholar] [CrossRef]
- Stanford, P.K. Scientific realism, the atomic theory, and the catch-all hypothesis: Can we test fundamental theories against all serious alternatives. Br. J. Philos. Sci. 2009, 60, 253–269. [Google Scholar] [CrossRef]
- Stanford, P.K. Exceeding Our Grasp. Science, History, and the Problem of Unconceived Alternatives; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Shih, Y.-L.; Rothfield, L. The bacterial cytoskeleton. Microbiol. Mol. Rev. 2006, 70, 729–754. [Google Scholar]
- Ingerson-Mahar, M.; Gitai, Z. A growing family: The expanding universe of the bacterial cytoskeleton. FEMS Microbiol. Rev. 2012, 36, 256–266. [Google Scholar]
- Bonduriansky, R.; Day, T. Extended Heredity: A New Understanding of Inheritance and Evolution; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Bonduriansky, R. Rethinking heredity, again. Trends Ecol. Evol. 2012, 27, 330–336. [Google Scholar] [CrossRef]
- Bonduriansky, R.; Day, T. Nongenetic inheritance and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 103–125. [Google Scholar] [CrossRef]
- Pigliucci, M. Do we need an extended evolutionary synthesis? Evolution 2007, 61, 2743–2749. [Google Scholar] [CrossRef]
- Danchin, E.; Charmantier, A.; Champagne, F.A.; Mesoudi, A.; Pujol, B.; Blanchet, S. Beyond DNA: Integrating inclusive inheritance into an extended theory of evolution. Nat. Rev. Genet. 2011, 12, 475–486. [Google Scholar] [CrossRef]
- Laland, K.N.; Uller, T.; Feldman, M.W.; Sterelny, K.; Müller, G.B.; Moczek, A.; Jablonka, E.; Odling-Smee, J. The extended evolutionary synthesis: Its structure, assumptions and predictions. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151019. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Genomes and Eukaryotic Phylogeny. In Organelles, Genomes and Eukaryote Phylogeny: An Evolutionary Synthesis in the Age of Genomics; Hirt, R.P., Horner, D.S., Eds.; Taylor & Francis: London, UK, 2004; pp. 335–351. [Google Scholar]
- Jablonka, E.; Lamb, M.J. Evolution in Four Dimensions. Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life; MIT Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Jablonka, E.; Lamb, M.J. The evolution of information in the major transitions. J. Theor. Biol. 2006, 239, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Lamb, M.J. Transgenerational Epigenetic Inheritance. In Evolution—The Extended Synthesis; Pigliucci, M., Müller, G.B., Eds.; MIT Press: Cambridge, MA, USA, 2010; pp. 135–174. [Google Scholar]
- Martinez Arias, A. Master Builder. How the New Science of the Cell Is Rewriting the Story of Life; Basic Books: London, UK, 2023. [Google Scholar]
- Müller, G.A.; Müller, T.D. (Patho)Physiology of glycosylphosphatidylinositol-anchored proteins II: Intercellular transfer of matter (inheritance?) that matters. Biomolecules 2023, 13, 994. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. The meaning of the cellular release of glycosylphosphatidylinositol-anchored proteins—Potential use as biomarkers. J. Nanomed. Res. 2018, 7, 203–219. [Google Scholar]
- Harraway, D.J. Crystals, Fabrics, and Fields. Metaphors that Shape Embryos; North Atlantic Books: Berkeley, CA, USA, 1976. [Google Scholar]
- Harraway, D. Die Neuerfindung der Natur. Primaten, Cyborgs und Frauen; Campus: Frankfurt am Main, Germany, 1995. [Google Scholar]
- Latour, B.; Woolgar, S. Laboratory Life. The Social Construction of Scientific Facts; Sage: London, UK, 1979. [Google Scholar]
- Latour, B. On actor-network theory. A few clarifications. Soz. Welt 1996, 47, 369–381. [Google Scholar]
- Law, J. After Method: Mess in Social Science Research; Routledge: London, UK, 2004. [Google Scholar]
- Barad, K. Meeting the Universe Halfway. Quantum Physics and the Entanglement of Matter and Meaning; Duke University Press: Durham, NC, USA, 2007. [Google Scholar]
- Barad, K. How material-discursive practices matter. Signs J. Women Cult. Soc. 2003, 28, 803–831. [Google Scholar] [CrossRef]
- Barad, K. Diffracting diffraction. Cutting together-apart. Parallax 2014, 20, 168–187. [Google Scholar] [CrossRef]
- Harwood, J. Styles of Scientific Thought: The German Genetics Community 1900–1933; University of Chicago Press: Chicago, IL, USA; London, UK, 1983. [Google Scholar]
- Harwood, J. Gesellschaftsstruktur als Analogie: Genetiker stellen sich die Zelle vor. Ber. Wissenschaftsgesch. 1989, 12, 157–158. [Google Scholar] [CrossRef]
- Kay, L.E. Who Wrote the Book of Life? A History of the Genetic Code; Stanford University Press: Redwood City, CA, USA, 2000. [Google Scholar]
- Fox-Keller, E.F. Refiguring Life: Metaphors of Twentieth-Century Biology; Columbia University Press: New York, NY, USA, 1995. [Google Scholar]
- Müller, G.A. Donation and acceptance in biological inheritance: The long path from Darwin’s gemmules, DNA and membranes to uniqueness and kinship. Adv. Hist. Stud. 2024, 13, 26–72. [Google Scholar] [CrossRef]
- Müller, G.A. “Centric” and “excluding” conceptions of biological inheritance. Adv. Hist. Stud. 2024, 13, 122–155. [Google Scholar] [CrossRef]
- Wilson, E.B. The Cell in Development and Heredity; Macmillan: New York, NY, USA, 1896. [Google Scholar]
- Wilson, E.B. An Atlas of the Fertilization and Karyokinesis of the Ovum; Macmillan: New York, NY, USA, 1895. [Google Scholar]
- Haldane, J.B.S. The Biochemistry of the Individual. In Perspectives in Biochemistry; Needham, J., Green, D.E., Eds.; Cambridge University Press: Cambridge, UK, 1937. [Google Scholar]
- Fleck, L. Entstehung und Entwicklung Einer Wissenschaftlichen Tatsache. Einführung in die Lehre vom Denkstil und Denkkollektiv; Suhrkamp: Frankfurt am Main, Germany, 1980. [Google Scholar]
- Fleck, L. Denkstile und Tatsachen; Suhrkamp: Berlin, Germany, 2011. [Google Scholar]
- Wheeler, M.; Clark, A. Genetic representation. Reconciling content and causal complexity. Br. J. Philos. Sci. 1999, 50, 103–135. [Google Scholar] [CrossRef]
- Szathmary, E. The evolution of replicatory. Philos. Trans. R. Soc. B 2000, 355, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Sterelny, K. Niche Construction, Developmental Systems, and the Extended Replicator. In Cycles of Contingency: Developmental Systems and Evolution; Oyama, S., Griffith, P.E., Gray, R.D., Eds.; MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Graham, J.M. Membrane Analysis; Bios Scientific Publishers Limited, Taylor & Francis: Milton Park, UK, 1997. [Google Scholar]
- Mokranjac, D.; Neupert, W. Thirty years of protein translocation into mitochondria: Unexpectedly complex and still puzzling. Biochim. Biophys. Acta 2009, 1793, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, N. Inclusion and exclusion in the history of developmental biology. Development 2019, 146, dev175448. [Google Scholar] [CrossRef]
- Jasanoff, S. Designs on Nature: Science and Democracy in Europe and the United States; Princeton University Press: Princeton, NJ, USA, 2005. [Google Scholar]
- Foucault, M. Geschichte der Gouvernementalität II. Die Geburt der Biopolitik; Suhrkamp: Frankfurt am Main, Germany, 2004. [Google Scholar]
- Comfort, N. The Science of Human Perfection. How Genes Became the Heart of American Medicine; Yale University Press: New Haven, CT, USA, 2012. [Google Scholar]
- Blumenberg, H. Schriften zur Technik; Suhrkamp: Berlin, Germany, 2015. [Google Scholar]
- Wuchterl, K. Kontingenz oder das Andere der Vernunft. Zum Verhältnis von Philosophie, Naturwissenschaft und Religion; Franz Steiner: Stuttgart, Germany, 2011. [Google Scholar]
- Dawkins, R. The Selfish Gene; Oxford University Press: Oxford, UK, 1976. [Google Scholar]
- Weismann, A. Die Continuität des Keimplasma’s als Grundlage Einer Theorie der Vererbung; Fischer: Jena, Germany, 1885. [Google Scholar]
- Flusser, V. Kommunikologie; Fischer: Frankfurt am Main, Germany, 2000. [Google Scholar]
- Battran, M. Jean-Baptiste de Lamarck (1744–1829) und 150 Jahre ‘Lamarckismus‘. Zur Geschichte Entwicklungsphysiologisch Orientierten Evolutionsdenkens; Franz Steiner: Stuttgart, Germany, 2023; Volumes 1 and 2. [Google Scholar]
- Landman, O.E. Inheritance of acquired characteristic revisited. BioScience 2022, 43, 696–705. [Google Scholar] [CrossRef]
- Kammerer, P. Vererbung erzwungener Fortpflanzungsanpassungen I. und II. Mitteilung: Die Nachkommen der spätgebornen Salamandra maculosa und der frühgebornen Salamandra atra. Arch. Entwicklungmech. Organ. 1907, XXV, 7–51. [Google Scholar] [CrossRef]
- Kammerer, P. Über Vererbung erworbener Eigenschaften. Das Wissen Alle 1908, 14, 6–216. [Google Scholar]
- Lysenko, T.D. Agrobiologie. Arbeiten über Fragen der Genetik, der Züchtung und des Samenbaus. 10. Beiheft zur “Sowjetwissenschaft”; Verlag Kultur und Fortschritt: Berlin, Germany, 1951. [Google Scholar]
- Lysenko, T.D. The influence of the time of the second autumn sowing on the conversion of spring wheat to winter wheat. Trudy Inst. Genet. 1958, 24, 232–237. [Google Scholar]
- Tollefsbol, T.O. Handbook of Epigenetics: The New Molecular and Medical Genetics; Academic Press: London, UK; Oxford, UK; Boston, MA, USA, 2011. [Google Scholar]
- Lickliter, R.; Witherington, D.C. Towards a truly developmental epigenetics. Hum. Dev. 2017, 60, 124–138. [Google Scholar]
- Halfmann, R.; Lindquist, S. Epigenetics in the extreme: Prions and the inheritance of environmentally acquired traits. Science 2010, 330, 629–632. [Google Scholar] [CrossRef]
- Hayes, W. Genetic transformation: A retrospective appreciation. First Griffith memorial lecture. J. Gen. Microbiol. 1966, 45, 385–397. [Google Scholar] [CrossRef]
- Judson, H.F. The Eight Day of Creation: The Makers of the Revolution in Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 1996. [Google Scholar]
- Wright, H.D. Obituary. William McDonald Scott and Frederick Griffith. Lancet 1941, 23, 588–589. [Google Scholar]
- Hopkins, F.G. The influence of chemical thought on biology. Lecture at Harvard tercentenary conference on Arts and Sciences. Science 1936, 84, 258. [Google Scholar]
- Hopkins, F.G. Biological Thought and Chemical Thought. A Plea for Unification. Linacre Lecture 1938. In Hopkins and Biochemistry, 1861–1947; Needham, J., Baldwin, E., Eds.; W. Heffer and Sons: Cambridge, MA, USA, 1949. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Model | Fractionation | Analysis | Detection | Author | Transferred Entities | Transferred Principle | Theory of Inheritance | Inherited Phenotype | Intention |
|---|---|---|---|---|---|---|---|---|---|---|
| Griffith 1.4–1.X | Homogenate | - Phospholipase - Centrifugation (high speed) | - Resistance - Auxotrophy | - Selection | tbd | - Prions - Int Disord Prot - OM-Vesicles - Micelle-like LPS Compl. | - Substance/Matter | - Including - Holistic | - Minor Features - Small Differences | - Mode of Inheritance |
| Griffith 1.3 | - Chloroform - Centrifugation - Nuclease - Protease - Water-Sol. Fr. | - Virulence | -Microscopy - Serotypes | Avery MacLeod McCarty Alloway | - DNA | - Shape/Information | - Centric | - Novel Cells | ||
| Griffith 1.2 | - Heat - Soluble Fract. | - Serotypes | Dawson | + | - Excluding | - Matter of Inheritance | ||||
| Griffith 1.1 | Mouse | - Heat | Griffith Neufeld Levinthal | - Substance/Matter | - “Hard“ | |||||
| Griffith 1.0 | - none | - Lethality Mice | Griffith | - Bacterial Variability - Infections |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.A. The Transformation Experiment of Frederick Griffith I: Its Narrowing and Potential for the Creation of Novel Microorganisms. Bioengineering 2025, 12, 324. https://doi.org/10.3390/bioengineering12030324
Müller GA. The Transformation Experiment of Frederick Griffith I: Its Narrowing and Potential for the Creation of Novel Microorganisms. Bioengineering. 2025; 12(3):324. https://doi.org/10.3390/bioengineering12030324
Chicago/Turabian StyleMüller, Günter A. 2025. "The Transformation Experiment of Frederick Griffith I: Its Narrowing and Potential for the Creation of Novel Microorganisms" Bioengineering 12, no. 3: 324. https://doi.org/10.3390/bioengineering12030324
APA StyleMüller, G. A. (2025). The Transformation Experiment of Frederick Griffith I: Its Narrowing and Potential for the Creation of Novel Microorganisms. Bioengineering, 12(3), 324. https://doi.org/10.3390/bioengineering12030324