Simple Summary
Immunotherapy for the treatment of cancer seeks to use the body’s own immune system to recognize and eliminate tumor cells. Some of the most successful immunotherapies in human medicine have relied on the generation of biological reagents specific to humans, such as tumor antigen vaccines and humanized monoclonal antibodies. Veterinarians would like to incorporate immunotherapies into oncogenic medicine; however, the readily available reagents are biological therapies designed for human use, and their utility in veterinary medicine remains unknown. In some instances, such as tumor antigen vaccination, humanized reagents may prove advantageous in animal species. In other instances, such as the use of humanized monoclonal antibodies, the treatment may fail as a result of the animal’s own immune system rejecting the human reagent. Here, we review the potential use of these reagents for veterinary oncology and explore other possible reagents that may have “universal” applicability in different animal species.
Abstract
The emergence of immunotherapy for the treatment of human cancers has heralded a new era in oncology, one that is making its way into the veterinary clinic. As the immune system of many animal species commonly seen by veterinarians is similar to humans, there is great hope for the translation of human therapies into veterinary oncology. The simplest approach for veterinarians would be to adopt existing reagents that have been developed for human medicine, due to the potential of reduced cost and the time it takes to develop a new drug. However, this strategy may not always prove to be effective and safe with regard to certain drug platforms. Here, we review current therapeutic strategies that could exploit human reagents in veterinary medicine and also those therapies which may prove detrimental when human-specific biological molecules are used in veterinary oncology. In keeping with a One Health framework, we also discuss the potential use of single-domain antibodies (sdAbs) derived from camelid species (also known as Nanobodies™) for therapies targeting multiple veterinary animal patients without the need for species-specific reformulation. Such reagents would not only benefit the health of our veterinary species but could also guide human medicine by studying the effects of outbred animals that develop spontaneous tumors, a more relevant model of human diseases compared to traditional laboratory rodent models.
1. Introduction
Veterinary medicine often adopts therapeutic strategies developed for human medicine to treat similar diseases in animal patients. These treatments often include medications approved for use in humans that target similar physiological pathways in animals. For example, the most common agents used for the treatment of veterinary oncological diseases are cytotoxic chemotherapies, such as alkylating agents (e.g., cyclophosphamaide), vinca alkaloids (e.g., Vincristine), anthracyclines (e.g., Doxorubicin) and platinum-based drugs (e.g., Carboplatin), which were originally developed for human cancer patients. The strategy has shown benefit to the veterinary profession, as bypassing novel drug development saves both time and funding. While this strategy may prove successful when using small (or even large) pharmaceutical chemicals, it is more problematic when considering the use of biological agents, which are often tailored specifically for the use in a single species. Nowhere is this more important than in the recently emerging field of tumor immunotherapy, which relies on activating or reinvigorating the patient’s own immune system to recognize and remove transformed cells while sparing healthy tissue. Mostly developed for use in human medicine, the principles involved in immunotherapy are expected to translate into veterinary medicine []. However, the currently existing reagents themselves may or may not yield the desired results. It, therefore, behooves us to consider when and where human biological reagents can be adapted directly to the veterinary clinic and when they should be avoided.
2. Tumor Immunotherapy: General Principles
The goal of using the body’s own immune system to destroy transformed cells is over a century old; however, successfully harnessing the immune system to fight cancer has remained elusive []. In their seminal 1957 paper, Prehn and Main began by stating “The history of attempts to immunize against cancer is one of long frustration []”, a phrase which, until the last decade or so, could have been the introductory sentence to numerous PhD theses on tumor immunology. Since then, immunotherapy has moved from a once unattainable goal to the first-line treatment option for a growing number of specific cancers [].
What has changed in recent years to spur on this paradigm shift in cancer treatment? There have been several important discoveries made, which demonstrate why immunotherapy can be effective. Tumors are derived from “self” tissues; thus, the prior prevailing thought in the field was that tumor reactive T-cells would be deleted by the positive thymic selection of lymphocytes in order to reduce the risk of autoimmune reactions. However, the thinking in the field shifted, as it became known that tumor cells can harbor specific antigens that the immune system can target. There are several classes of these tumor-associated antigens [], including overexpressed self-proteins [,] and unique new antigens, known as neoantigens, created as a result of continuous mutations, which occur during gene reorganization and tumor progression [,]. Neoantigens, in particular, are attractive targets for the immune system, as T-cells can be capable of recognizing such antigens that make the tumor appear foreign or non-self. The presence of neoantigens and other associated tumor antigens has spurred on the development of tumor antigen vaccines, which could be used to therapeutically treat oncologic diseases [,]. Therefore, even though they are derived from self-tissues, tumor cells can, in fact, harbor the antigens that mark them as targets for the adaptive immune response.
Going hand-in-hand with the continuous discovery of tumor antigens is the increased appreciation for cytotoxic T-cells in eliminating cancer cells [,]. Cytotoxic T-cells are responsible for eliminating self-cells that have become infected or transformed. To accomplish this, T-cells must be able to recognize that a cell in the body is diseased, a process which involves the presentation of disease-derived peptides on major histocompatibility complex (MHC) class I molecules. In addition to detecting a disease-associated peptide, cytotoxic T-cells also receive secondary signals from target cells and the target cell’s environment, which can further activate or, alternatively, inhibit T-cell-mediated killing []. Negative signals, such as checkpoint signals, delivered to T-cells are necessary in order to control the immune response and prevent an unnecessary or potentially dangerous autoimmune response; however, tumors often hijack the expression of checkpoint molecules to thwart cytotoxic T-cell responses [,]. Blocking these checkpoint signaling pathways is, therefore, an attractive way to overcome a T-cell-suppressive environment in the tumor and reestablish tumor-specific cytotoxic T-cell responses against cancer cells.
These findings provided the drive to push immunologists and clinicians to develop and test new ways to boost anti-tumor immune responses in the clinic, with impressive results. Numerous strategies have been adopted to prime and enhance the adaptive immune system’s ability to recognize and kill tumor cells, such as vaccination with tumor-specific antigens [], in vitro expansion and reintroduction of immune cells capable of supporting anti-tumor responses [], development of monoclonal antibodies, which can directly target tumor cells for destruction [], genetically engineering autologous T-cells to react with tumor-specific proteins on the cell surface [] and the development of monoclonal antibody therapies, which can block negative signals delivered from the tumor microenvironment to T-cells []. As a result, immunotherapy is emerging to be the first-line option for oncologists [].
Veterinary oncologists, looking at the success of human immunotherapy, will adopt some of these strategies for animal patients. Some treatments, such as the expansion of autologous patient immune cells or the generation of chimeric-antigen receptor (CAR) T-cells, will likely be too expensive for clinical applications, at least in the near future. Treatment options that are not cell-based therapies offer a more cost-effective and simpler option; however, many of the reagents used in immunotherapy are biological molecules and are developed specifically for use in humans. Considerations of how biologic-based reagents will work in a different species will be needed before this existing suite of therapeutic products developed for humans can be used safely and effectively in the veterinary clinic. Below, we will review some of the existing products in human medicine and describe which therapies may have advantages in veterinary species and which may not.
3. Human Tumor Antigen Vaccination
One branch of immunotherapy incorporates our understanding of tumor-associated antigens to develop vaccines, which target said antigens. Tumor antigens could be introduced to the patient using multiple strategies, such as vaccination with purified proteins or peptides mixed with adjuvants, using recombinant attenuated pathogens to deliver antigens or by the introduction of nucleic acid sequences encoding the tumor antigen []. The recent successes obtained through the use of mRNA vaccines to drive protective immune responses against SARS-CoV-2 across the globe have demonstrated the utility in the use of nucleic-acid-based vaccines to successfully prime the immune system to recognize the introduced antigen. It is, therefore, plausible that we will see more and more nucleic acid vaccines targeting tumor antigens in the future.
The orthologs of several human tumor-associated antigens have also been identified in companion animals, and these proteins are often present in similar tumor types as in humans. Many of these potential targets can be found in melanomas, which is important as canine melanomas are a model for human melanomas []. Bergman and colleagues set out to determine if tyrosinase could be an immune target in canine melanoma []. To test this, dogs were vaccinated with a readily available human DNA plasmid vaccine that encodes human tyrosinase (hTyr). The amino acid similarity between hTyr and canine Tyr is 91%, which suggests that the differences in amino acid sequences could overcome any adaptive immune tolerance []. Initial clinical studies demonstrated potential benefits, including extended survival times and the induction of anti-tyrosinase antibodies, which suggest that a xeongenic tumor-associated antigen could serve as a target to generate clinically effective anti-tumor T-cell responses [,,].
Several clinical trials and retrospective studies were subsequently conducted with the DNA-based hTyr vaccine, sold under the name Oncept®, for the treatment of melanoma [,,,,,]. Some studies showed a relative degree of efficacy [,], though it should be noted that others did not show a benefit []. Most of these studies were retrospective studies and may not accurately capture the true impact of a drug treatment [,,]. Nevertheless, Oncept is safe and well-tolerated, and as future studies are conducted, it is likely that the effect of prognostic indicators, such as tumor size and lymph node metastasis, on Oncept efficacy will be better detailed []. A recent case report also reported that Oncept can be combined with a tyrosine kinase inhibitor to treat lingual metastatic melanoma [], raising the exciting possibility of a combinational approach with a vaccine and other adjuvant therapies, including chemotherapy, to elicit better clinical outcomes. Therefore, xenogenic vaccination with DNA-encoded plasmids is a potential approach for using existing therapies developed for humans in a veterinary oncology setting.
Other melanoma-associated tumor antigens could also be the target of xenogenic vaccination. Chondroitin sulfate proteoglycan-4 (CSPG-4) is another potential antigenic target in multiple tumor types []. Human CSPG-4 is very similar to canine CSPG-4 (82% homology and 88% similarity) and would, therefore, make an excellent choice of antigens []. A DNA-based vaccine encoding hCSPG-4 has been used in multiple clinic trials to induce adaptive immune responses against cCSPG-4 in dogs with melanoma, and the initial results appear promising [,,]. Riccardo et al. tested hCSPG-4 vaccination in dogs with stage II-III oral malignant melanoma and noted extended 6- and 12-month survival times following surgical resection and vaccination compared to animals who received surgery alone, accompanied by an increase in anti-CSPG-4 antibodies in serum []. The authors used a technique referred to as electrovaccination, where, immediately following the injection of plasmid DNA into the muscle, a series of short electrical pulses were applied to the injection site to boost plasmid DNA uptake into the nucleus of cells []. A second follow-up study, by Piras et al., confirmed the extended survival times and reduced lung metastasis in vaccinated dogs compared to unvaccinated controls []. In addition, a retrospective study also suggested that vaccination with hCSPG-4 can increase survival time, especially when initial surgery focused on curative intent rather than cytoreduction only []. Taken together, these studies suggest that xenogeneic vaccination with hCSPG-4 in canine patients following surgical removal of melanoma tumors can enhance survival time by possibly driving an anti-tumor CSPG-4-specific response [,,].
Xenogeneic DNA vaccines are not the only method to introduce human antigenic proteins into veterinary patients. Recombinant pathogens, such as viruses and bacteria, can be engineered to express antigenic proteins when infecting a host, often driving impressive immune responses [,]. Recent studies have demonstrated the potential efficacy of using recombinant Listeria monocytogenes bacteria to deliver tumor antigens to hosts []. Due to the nature of the bacteria, L. monocytogenes infections result in the secretion of bacterial antigens into the host cell cytoplasm, which are subsequently presented by MHC class I molecules in a highly efficient manner [,]. There are currently several human clinical studies investigating the use of Lm-vaccines to induce anti-tumor immune responses [].
In veterinary species, an Lm-based vaccine for the treatment of osteosarcoma (OSA) is currently in clinical trials []. Osteosarcoma is the most common bone neoplasia in dogs, accounting for 85% of bone tumors [], and is thought to have immunogenic tumor properties [,,]. When local disease is controlled, pulmonary metastasis is thought to be the biggest contributor to life-limiting disease processes [,]. Canine osteosarcoma has many similarities with human osteosarcoma [,,,,], as it is the most common bone tumor and commonly affects larger breeds and taller individuals [,]. Additionally, the proto-oncogene erbB-2, also known as Her-2, has been found to be overexpressed in canine osteosarcoma, similar to human osteosarcoma []. Thus, canine osteosarcoma disease is of interest not only for therapeutic treatments in companion animals but also for translation purposes in human oncology. Therapeutic improvements in both the human and veterinary clinics for long-term treatment options are needed, as the current standard of care has not changed in over 20 years. Immunotherapy options are promising but have yet to be fully understood [].
ADXS31-164 is an Lm-based vaccine vector expressing human Her-2 that was developed originally for use in human patients [] but is now also being considered for use in canine patients with osteosarcoma []. In a clinical trial, enrolling 18 dogs that had a primary tumor removed via amputation and received chemotherapy, ADXS31-164 administration resulted in increased median survival time compared to historical controls []. Several of the dogs in the clinical trial also showed evidence of increased Her-2-specific T-cell responses following vaccination []. The vaccine is available in lyophilized form, and a recent study noted some adverse events in dogs receiving the vaccine [], although it should be noted that some animals tested positive for Listeria following vaccination [].
Combined, the data from these studies suggest a potential benefit for the use of a humanized agent in veterinary medicine. By using a human tumor-associated antigen, an adaptive immune response is generated that recognizes the orthologous animal antigen, perhaps even breaking previously existing immune tolerance to the targeted protein. It is likely that the slight differences between the human and animal proteins are enough to initiate an adaptive immune response, which, in the case of vaccination, is of benefit to the patient. However, as we will see in the next section, those same differences can lead to undesired immune responses to other therapies (see Figure 1).
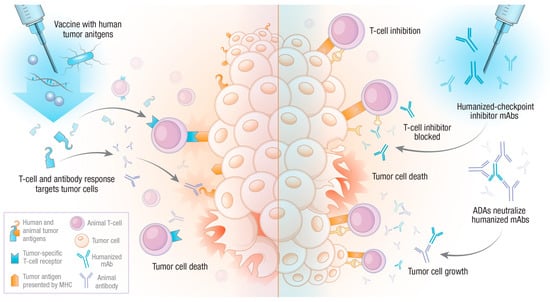
Figure 1.
Potential outcomes of humanized biological reagents in veterinary oncology. The administration of humanized biological compounds to veterinary patients will lead to an adaptive immune response against xenogenic reagents. In the case of tumor antigen vaccination (left side), the result may be deemed successful if the animal’s adaptive immune response can cross-react with the orthologous antigen or break tolerance. However, animal adaptive immune forces may also recognize humanized monoclonal antibodies, for instance, checkpoint inhibitors (right side). This can lead to anti-drug antibody (ADA) responses, which neutralize therapeutic antibodies, resulting in a T-cell inhibitory state, allowing for tumor growth.
4. Humanized Monoclonal Antibodies
Some of the most exciting results of human immunotherapies come from the use of checkpoint inhibitors. These reagents are monoclonal antibodies, which specifically block the interaction of suppressive signaling molecules on T-cells with their cognate ligands, ensuring that cytotoxic T-cells can be maintained in an active state and allowing for the killing of tumor cells []. To date, the most effective checkpoint inhibitor drug targets are CTLA-4, the PD-1/PD-L1 signaling pathways and LAG-3, which can be targeted by a recent FDA-approved drug, Relatlimab []. Relatlimab joins eight other monoclonal antibodies approved by the FDA to treat a variety of cancer types. The impact of these therapies on the field of oncology cannot be understated and is the subject of many excellent reviews [,,].
Orthologs of negative stimulators of T-cells are annotated in the genomes of many vertebrate species available in the NCBI database. Additionally, the expression of these proteins has been detected in the tumors of companion animals. For instance, PD-1/PD-L1 expression has been shown to be upregulated in a number of different canine tumors [,,,]. Monoclonal antibodies, which detect canine PD-1/PD-L1, have been developed [,,,], which could be useful for both diagnostic and therapeutic use in the veterinary clinic. Furthermore, blocking the PD-1 and PD-L1 axis in vitro with mAbs antibodies can enhance T-cell function [,,], as can blocking CTLA-4 []. Additionally, perhaps most excitingly, several FDA-approved checkpoint inhibitor antibodies can recognize and even block canine PD-1/PD-L1 interactions in vitro with atezolizumab (anti-PD-L1), showing the most robust increase in the production of the activation marker IFNγ []. Therefore, at least in dogs, and likely in cats [], negative T-cell signaling pathways exist, are present in tumors and can be targeted with monoclonal antibodies to enhance T-cell function. However, can the checkpoint inhibitor antibodies developed for human use be administered in other animals?
Antibodies have been used clinically for over 100 years, even before their identity was known and their functions were completely understood. Often, antibodies for use in human medicine were generated in other mammalian species. For instance, patients with diptheria were often treated with diptheria antitoxin (DAT), a polyclonal antibody solution derived from horses inoculated with Cornybacterium diphteriae, which neutralizes the diptheria toxin produced during infection []. However, the administration of DAT requires careful monitoring to be sure that patients do not develop hypersensitivity reactions []. The reader is encouraged to review Silverstein’s paper detailing Pirquet’s description of serum sickness from the early 1900s [], where Pirquet correctly identified the cause of serum sickness occurring when the patient recognizes the transferred antibody as a foreign agent, and the body mounts its own adaptive immune response to target the therapeutic antibody. The development of antibodies that recognize these exogenous therapeutic immunoglobulins are referred to as anti-drug antibodies (ADAs).
While the first descriptions of ADAs were in response to adoptively transferred polyclonal antibodies in sera, ADAs can also be detected in highly purified preparations of monoclonal antibodies. The first monoclonal antibody therapy approved for use in humans in the United States was muromonab CD3, a mouse monoclonal antibody targeting CD3 and widely used to suppress immune responses in transplant patients []. However, recipients often developed ADAs, which induced side effects [,]. Future generations of monoclonal antibody therapies partially solved this problem using the introduction of chimeric monoclonal antibodies: recombinant antibodies where most of the constant region of the mouse monoclonal antibody is replaced with a human Fc. However, even these chimeric antibodies still elicited ADAs. For instance, infliximab, a chimeric mAb that targets TNFα to prevent inflammatory reactions, is often immunogenic in patients. In fact, ADA responses can require an increasing dose of infliximab during treatment to maintain its efficacy [,,,]. In some instances, chimeric mAb can induce hypersensitivity responses. Cetuximab is a chimeric mAb that binds to the epithelial growth factor receptor (EGFR) on cancer cells to target the cells for destruction []. However, cetuximab treatment can result in hypersensitivity reactions in patients with an atopic history []. This has been linked to patient IgE antibodies, which can target a particular glycan, galactose-alpha-1,3-galactose (alpha-gal) [], a common antigenic target in patients who exhibit Alpha-gal Syndrome (AGS), where an intolerance to mammalian-derived food products develops [].
ADA responses generated following the adoptive transfer of fully murine or even chimeric antibodies can lower the efficacy of the drug, or worse, create or exacerbate hypersensitivity reactions [] in humans. Therefore, it is likely that the use of human monoclonal antibodies in veterinary patients will almost certainly induce similar ADA responses. ADA responses have been documented in dogs treated with monoclonal 231 (mAb231), a murine-based antibody therapy developed in the 1980s for treating lymphomas in dogs [,]. In human medicine, it has become increasingly apparent that there is a need to fully “humanize” monoclonal antibodies to reduce the likelihood of generating ADAs following treatment []. Many of the current immunotherapies in use consist of humanized antibodies, where only the amino acid sequence of the antigen-specific CDRs remain in the final product.
To circumvent the anti-drug antibody responses elicited in veterinary patients, antibody-based checkpoint inhibitor therapies will need to be re-engineered for each species of interest. The caninization of antibodies, where the constant domain of the (usually) rodent monoclonal antibody is replaced with the constant region of canine antibodies, has been reported. For instance, the anti-IL-31 antibody Lokivetmab, which is used in the treatment of atopic and allergic dermatitis [,,], is a fully caninized monoclonal antibody. Anti-nerve growth factor mAbs have also been adapted for both canine and feline patients [,,]. Fully caninized antibodies that could be used as checkpoint inhibitors are currently being developed. Recent pilot studies were conducted with caninized anti-PD-1 and anti-PD-L1 antibodies, which showed the caninized antibodies to be relatively safe [,]. Participant numbers were limited, but preliminary evidence also suggested a potential benefit to treatment with canonized anti PD-1 antibodies []. Studies with anti-PD-L1 antibodies suggest limited clinical benefit [,]. A caninized anti-CTLA-4 has also been developed and has been shown to enhance in vitro T-cell function []. While it is likely that caninized and felinized monoclonal antibodies will be developed in the future, a great deal of work will need to be undertaken to understand the functions and distributions of FcRs in each veterinary species of interest in order to successfully incorporate monoclonal antibody therapies. The speciation of checkpoint inhibitor antibodies will be of little use if these therapeutics trigger massive histamine release or activate the complement cascade upon binding to their target, which may occur if the wrong Fc portion of an antibody is coupled to a PD-1 or CTLA-4 targeting CDR.
Because of the potential for poorer target recognition, the triggering of anti-drug antibody responses and the lack of detailed knowledge regarding veterinary FcR usage and distribution, the development of checkpoint inhibitor therapy for veterinary species remains a daunting task. While future research should certainly attempt to tackle this problem, other treatment options may offer the advantage of increased safety to use in veterinary species.
5. Single-Domain Antibodies (sdAbs)
Studying the immune system in different animal species can often yield surprising and useful findings, perhaps none more so than the discovery of single-chain antibodies, first identified in camelid species [] and later in cartilaginous fish [], in an interesting case of divergent evolution. Camelid single-chain antibodies have captured the imagination of the biotech world, as they offer up a much simpler way to produce proteins with antibody-like specificity but through the production of a single protein, instead of both a heavy- and light-chain immunoglobulin [,]. Furthermore, a single immunoglobulin domain containing the CDR can be produced, which has the ability to recognize its target antigen but carries none of the remaining Fc region of the immunoglobulin. These molecules are known as single-domain antibodies (sdAbs) and often referred to as Nanobodies™. These reagents offer the ability to target a molecule with antibody-like specificity but without the potential downstream hazards of antibody therapies that can be related to FcR interactions. From a veterinarians’ perspective, single-domain antibodies hold great promise for the simple reason that they could be used in multiple species without the need for species-specific reformulation, as would be necessary for the more complex monoclonal antibodies. We will, therefore, describe, in more detail, the emerging literature regarding the use of sdAbs in the clinic, paying particular attention to their use in checkpoint inhibitor therapy (Figure 2).
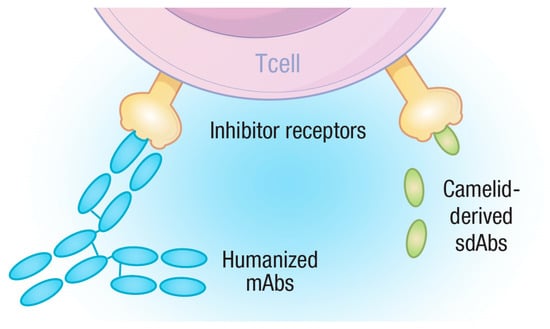
Figure 2.
Diagram depicting the rationale for sdAbs as checkpoint inhibitors. Monoclonal antibodies consist of two heterodimers containing multiple domains while sdAbs have a single domain. Both mAbs and sdAbs can recognize and bind to targets, such as T-cell inhibitory receptors, blocking their ability to negatively regulate T-cells, but sdAbs present far fewer epitopes for the adaptive immune system to target and are, therefore, less likely to be subjected to ADA responses.
5.1. The Advantage of Being Small
Immunoglobulin G (IgG) molecules, which make up nearly all classes of therapeutic monoclonal antibodies, are roughly ~140 kDa in size and consist of four proteins, two heavy-chain and two light-chain proteins, tethered together by several disulfide bonds. While IgG molecules are present in sera and can migrate into tissues, their relatively large size places limits on their utility. For instance, it has been known for decades that monoclonal antibodies poorly infuse solid tumors [,,,]. In contrast, sdAbs, which consist of a single immunoglobulin domain, are significantly smaller, with an average molecular weight of 15–16 kDa []. This size differential provides sdAbs with advantages over classical antibodies. The compact size of an sdAb allows it to access more compact bodily spaces, such as organs and solid tumors. When compared directly to conventional antibodies, sdAbs tend to accumulate in tumors with faster kinetics [,,]. Thus, the use of sdAbs will aid several hard-to-treat diseases compared to the more traditional antibody.
In addition to penetrating diverse tissues, the small size of sdAbs also allows for quicker clearance from the body following injection []. This reduces the chances of off-target effects of the sdAbs, which will be clinically important if an sdAb is being used to deliver radioactive compounds to visualize tumors or toxic payloads to kill cancer cells. Furthermore, the dose of sdAbs required to achieve a therapeutic outcome is less than more traditional antibodies. SdAbs may also enhance the tumor penetration of classic antibodies [].
Finally, the small size of an sdAb limits the number of antigenic targets that could be introduced through the introduction of a xenogenic protein. By eliminating nearly 85% of the amino acid sequence of a monoclonal antibody, none of which is necessary for interacting with the antigenic target of the antibody, we can eliminate most immune-induced anti-drug responses. Indeed, studies in humans suggest a limited immunogenicity of sdAbs, though it should be noted that the literature is rather sparse [,]. SdAbs in clinical trials can be taken up by dendritic cells but cause minimal activation []. Rossotti et al. expertly reviewed the recent clinical and pre-clinical reports, which collectively suggest that even when ADAs develop following sdAb infusion, there appears to be a limited loss of efficacy []. However, there are two notable exceptions [,], which suggest that the ADAs to sdAbs may preclude their use in the clinic. In a surprising and non-intuitive manner, the sdAbs in question were more “humanized” versions than other camelid-derived sdAbs used in the clinic [,]. This observation is vexing, and clearly future work is needed to understand the ADAs to sdAbs. Veterinary medicine could help fill this gap in knowledge by testing sdAbs in a variety of species to determine the extent of ADA responses to sdAbs.
5.2. Towards Clinical Applications
The time for sdAb use in many different clinical applications is fast approaching. Already, several clinical trials using sdAbs for the treatment of a variety of diseases are underway. For a comprehensive list of the most updated reviews on the subject, the reader is encouraged to see [,,]. The first clinical trials to utilize sdAbs targeted the Von Willebrand factor, marketed as caplacizumab. Caplacizumab is now routinely used to treat thrombotic thrombocytopenic purpura [,]. Several other sdAbs targeting immune molecules, such as TNFα, IL-17 and the IL6-R, are in clinical or preclinical development for the treatment of a diversity of diseases [,,].
The adaptation of sdAb technology as a checkpoint inhibitor has obvious importance in not only animal medicine but human medicine as well. Targeting CTLA-4 or PD-1 in solid tumors has remained challenging, but the use of an sdAb in place of an antibody may allow for greater drug permeability into solid tumors, promoting the cytotoxic T-cell killing of tumor cells. Several recent studies have identified potential sdAbs, which may act as checkpoint inhibitors, primarily the PD-1/PD-L1 axis [,,]. Thus, it is entirely plausible that checkpoint inhibitor sdAbs could supplement or even replace existing antibody-based therapies in the future [,,].
Perhaps most importantly, checkpoint inhibitor sdAbs might be readily transitioned into the veterinary clinic with limited modifications for the treatment of tumors in a variety of species. Not only would this greatly expand the arsenal of veterinary oncologists to treat patients but it could serve as a unique opportunity for comparative oncology studies. In addition to the previously discussed oral malignant melanoma and osteosarcoma, several cancers in companion animals are close mimics of human disease, such as canine multicentric lymphoma [,,], canine invasive urothelial cell carcinoma [,,,] and feline mammary carcinoma [,,]. In some instances, they are considered the best animal models for particular cancers. One could imagine clinical trials that combine checkpoint inhibitor therapy with more traditional chemotherapies in spontaneously occurring tumors in naturally outbred species, which closely mimic human disease. Not only would such trials benefit companion animal health but they could also provide very valuable information to inform human medicine, and they would occur at a fraction of the cost of human clinical trials. It is, therefore, imperative that the field not only develops checkpoint inhibitor sdAbs but also quickly ascertains their potential for use in veterinary species. Initial clinical trials in veterinary patients should not only determine any potential toxicity related to the administration of sdAbs but also determine if patients generate an ADA directed against asAbs.
6. In the Future
The promise of immunotherapy for the treatment of veterinary tumors is on the horizon; however, significant hurdles remain. Chief among them is the expense and time taken for drug development, which can often limit treatment options in animal species. Using off-the-shelf products intended for human use can eliminate some of these hurdles. In some instances, such as vaccination with human tumor antigens, the xenogenic differences will prove to be beneficial, as the slight differences between amino acid sequences in the target protein can successfully drive adaptive immune responses. However, this can be a double-edged sword as the same adaptive immune forces can target humanized reagents, such as checkpoint inhibitor antibodies, and, thus, induce immune-mediated anti-drug responses, which will, at best, eliminate drug efficacy and, at worst, lead to immune-induced hypersensitivity reactions. While it is possible to create species-specific checkpoint inhibitor monoclonal antibodies, a more exciting and useful approach may lie in the development of checkpoint inhibitor sdAbs, which are far more likely to be utilized in multiple species. The field needs to urgently explore this potentially groundbreaking therapy, which in a true One Health framework and will not only benefit animal health but can also teach us about human health.
Author Contributions
Conceptualization, J.N.H., C.E.R., D.V.M., S.B. and B.P.D.; writing—original draft preparation, J.N.H. and B.P.D.; writing—review and editing, J.N.H., C.E.R., D.V.M., S.B. and B.P.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded in part by the deLaubenfels Comparative Health Research and Education Fund of the Oregon State University Foundation and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI130059. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
DM and CR are founding members and co-owners of Biotesserae Inc. and did not receive any compensation or remuneration for their contributions to this work. Biotesserae did not provide any funding for this work.
References
- Klingemann, H. Immunotherapy for Dogs: Still Running behind Humans. Front. Immunol. 2021, 12, 665784. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Prehn, R.T.; Main, J.M. Immunity to methylcholanthrene-induced sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar]
- Segal, E.M. Immunotherapy in the frontline management of advanced and metastatic NSCLC. Am. J. Manag. Care 2021, 27, S323–S332. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Caballero, O.L.; Chen, Y.T. Cancer/testis (CT) antigens: Potential targets for immunotherapy. Cancer Sci. 2009, 100, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed oncogenic tumor-self antigens. Hum. Vaccines Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2018, 37, 173–200. [Google Scholar] [CrossRef]
- Bollineni, R.C.; Tran, T.T.; Lund-Johansen, F.; Olweus, J. Chasing neoantigens; invite naïve T cells to the party. Curr. Opin. Immunol. 2022, 75, 102172. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.T.; Street, S.E.A.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-Mediated Cytotoxicity Is Critical for Surveillance of Spontaneous Lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Bottomly, K. Signals and signs for lymphocyte responses. Cell 1994, 76, 275–285. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef]
- Zebley, C.C.; Youngblood, B. Mechanisms of T cell exhaustion guiding next-generation immunotherapy. Trends Cancer 2022, 8, 726–734. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Schuler, G.; Schuler-Thurner, B.; Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. [Google Scholar] [CrossRef]
- Sun, Z.; Fu, Y.X.; Peng, H. Targeting tumor cells with antibodies enhances anti-tumor immunity. Biophys. Rep. 2018, 4, 243–253. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.M.; Berzofsky, J.A. Strategies for developing and optimizing cancer vaccines. F1000Research 2019, 8, 654. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Adissu, H.A.; Wei, B.-R.; Michael, H.T.; Merlino, G.; Simpson, R.M. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J.; McKnight, J.; Novosad, A.; Charney, S.; Farrelly, J.; Craft, D.; Wulderk, M.; Jeffers, Y.; Sadelain, M.; Hohenhaus, A.E.; et al. Long-Term Survival of Dogs with Advanced Malignant Melanoma after DNA Vaccination with Xenogeneic Human Tyrosinase: A Phase I Trial1. Clin. Cancer Res. 2003, 9, 1284–1290. [Google Scholar] [PubMed]
- Liao, J.C.; Gregor, P.; Wolchok, J.D.; Orlandi, F.; Craft, D.; Leung, C.; Houghton, A.N.; Bergman, P.J. Vaccination with human tyrosinase DNA induces antibody responses in dogs with advanced melanoma. Cancer Immun. 2006, 6, 8. [Google Scholar]
- Bergman, P.J.; Camps-Palau, M.A.; McKnight, J.A.; Leibman, N.F.; Craft, D.M.; Leung, C.; Liao, J.; Riviere, I.; Sadelain, M.; Hohenhaus, A.E.; et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine 2006, 24, 4582–4585. [Google Scholar] [CrossRef]
- Grosenbaugh, D.A.; Leard, A.T.; Bergman, P.J.; Klein, M.K.; Meleo, K.; Susaneck, S.; Hess, P.R.; Jankowski, M.K.; Jones, P.D.; Leibman, N.F.; et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am. J. Vet. Res. 2011, 72, 1631–1638. [Google Scholar] [CrossRef]
- McLean, J.L.; Lobetti, R.G. Use of the melanoma vaccine in 38 dogs: The South African experience. J. S. Afr. Vet. Assoc. 2015, 86, 1246. [Google Scholar] [CrossRef]
- Ottnod, J.M.; Smedley, R.C.; Walshaw, R.; Hauptman, J.G.; Kiupel, M.; Obradovich, J.E. A retrospective analysis of the efficacy of Oncept vaccine for the adjunct treatment of canine oral malignant melanoma. Vet. Comp. Oncol. 2013, 11, 219–229. [Google Scholar] [CrossRef]
- Treggiari, E.; Grant, J.P.; North, S.M. A retrospective review of outcome and survival following surgery and adjuvant xenogeneic DNA vaccination in 32 dogs with oral malignant melanoma. J. Vet. Med. Sci. 2016, 78, 845–850. [Google Scholar] [CrossRef]
- Verganti, S.; Berlato, D.; Blackwood, L.; Amores-Fuster, I.; Polton, G.A.; Elders, R.; Doyle, R.; Taylor, A.; Murphy, S. Use of Oncept melanoma vaccine in 69 canine oral malignant melanomas in the UK. J. Small Anim. Pract. 2017, 58, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Turek, M.; LaDue, T.; Looper, J.; Nagata, K.; Shiomitsu, K.; Keyerleber, M.; Buchholz, J.; Gieger, T.; Hetzel, S. Multimodality treatment including ONCEPT for canine oral melanoma: A retrospective analysis of 131 dogs. Vet. Radiol. Ultrasound 2020, 61, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Hayes, A.; Schiavo, L.; Dobson, J. Multimodal Treatment of a Canine Lingual Melanoma Using a Combination of Immunotherapy and a Tyrosine Kinase Inhibitors. Vet. Sci. 2022, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential as an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2018, 8, 01911. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Iussich, S.; Maniscalco, L.; Lorda Mayayo, S.; La Rosa, G.; Arigoni, M.; De Maria, R.; Gattino, F.; Lanzardo, S.; Lardone, E.; et al. CSPG4-Specific Immunity and Survival Prolongation in Dogs with Oral Malignant Melanoma Immunized with Human CSPG4 DNA. Clin. Cancer Res. 2014, 20, 3753–3762. [Google Scholar] [CrossRef]
- Piras, L.A.; Riccardo, F.; Iussich, S.; Maniscalco, L.; Gattino, F.; Martano, M.; Morello, E.; Lorda Mayayo, S.; Rolih, V.; Garavaglia, F.; et al. Prolongation of survival of dogs with oral malignant melanoma treated by en bloc surgical resection and adjuvant CSPG4-antigen electrovaccination. Vet. Comp. Oncol. 2017, 15, 996–1013. [Google Scholar] [CrossRef]
- Giacobino, D.; Camerino, M.; Riccardo, F.; Cavallo, F.; Tarone, L.; Martano, M.; Dentini, A.; Iussich, S.; Lardone, E.; Franci, P.; et al. Difference in outcome between curative intent vs marginal excision as a first treatment in dogs with oral malignant melanoma and the impact of adjuvant CSPG4-DNA electrovaccination: A retrospective study on 155 cases. Vet. Comp. Oncol. 2021, 19, 651–660. [Google Scholar] [CrossRef]
- Sardesai, N.Y.; Weiner, D.B. Electroporation delivery of DNA vaccines: Prospects for success. Curr. Opin. Immunol. 2011, 23, 421–429. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Solovyeva, V.V.; Kitaeva, K.V.; Dunham, S.P.; Khaiboullina, S.F.; Rizvanov, A.A. Recombinant Viruses for Cancer Therapy. Biomedicines 2018, 6, 94. [Google Scholar] [CrossRef]
- Oladejo, M.; Paterson, Y.; Wood, L.M. Clinical Experience and Recent Advances in the Development of Listeria-Based Tumor Immunotherapies. Front. Immunol. 2021, 12, 642316. [Google Scholar] [CrossRef]
- Wolf, B.J.; Princiotta, M.F. Processing of Recombinant Listeria monocytogenes Proteins for MHC Class I Presentation Follows a Dedicated, High-Efficiency Pathway. J. Immunol. 2013, 190, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.S.; Sijts, A.J.; Pamer, E.G. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J. Immunol. 1995, 155, 5227–5233. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.J.; Gnanandarajah, J.S.; Engiles, J.B.; Gray, F.; Laughlin, D.; Gaurnier-Hausser, A.; Wallecha, A.; Huebner, M.; Paterson, Y. Immunotherapy with a HER2-Targeting Listeria Induces HER2-Specific Immunity and Demonstrates Potential Therapeutic Effects in a Phase I Trial in Canine Osteosarcoma. Clin. Cancer Res. 2016, 22, 4380–4390. [Google Scholar] [CrossRef]
- Ehrhart, N.P.; Ryan, S.D.; Fan, T.M. 24—Tumors of the Skeletal System. In Withrow and MacEwen’s Small Animal Clinical Oncology, 5th ed.; Withrow, S.J., Vail, D.M., Page, R.L., Eds.; W.B. Saunders: Saint Louis, MO, USA, 2013; pp. 463–503. [Google Scholar] [CrossRef]
- Brady, J.V.; Troyer, R.M.; Ramsey, S.A.; Leeper, H.; Yang, L.; Maier, C.S.; Goodall, C.P.; Ruby, C.E.; Albarqi, H.A.M.; Taratula, O.; et al. A Preliminary Proteomic Investigation of Circulating Exosomes and Discovery of Biomarkers Associated with the Progression of Osteosarcoma in a Clinical Model of Spontaneous Disease. Transl. Oncol. 2018, 11, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Troyer, R.M.; Ruby, C.E.; Goodall, C.P.; Yang, L.; Maier, C.S.; Albarqi, H.A.; Brady, J.V.; Bathke, K.; Taratula, O.; Mourich, D.; et al. Exosomes from Osteosarcoma and normal osteoblast differ in proteomic cargo and immunomodulatory effects on T cells. Exp. Cell Res. 2017, 358, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Wycislo, K.L.; Fan, T.M. The Immunotherapy of Canine Osteosarcoma: A Historical and Systematic Review. J. Vet. Intern. Med. 2015, 29, 759–769. [Google Scholar] [CrossRef]
- Wilk, S.S.; Zabielska-Koczywąs, K.A. Molecular Mechanisms of Canine Osteosarcoma Metastasis. Int. J. Mol. Sci. 2021, 22, 3639. [Google Scholar] [CrossRef]
- Selmic, L.E.; Burton, J.H.; Thamm, D.H.; Withrow, S.J.; Lana, S.E. Comparison of Carboplatin and Doxorubicin-Based Chemotherapy Protocols in 470 Dogs after Amputation for Treatment of Appendicular Osteosarcoma. J. Vet. Intern. Med. 2014, 28, 554–563. [Google Scholar] [CrossRef]
- Angstadt, A.Y.; Motsinger-Reif, A.; Thomas, R.; Kisseberth, W.C.; Guillermo Couto, C.; Duval, D.L.; Nielsen, D.M.; Modiano, J.F.; Breen, M. Characterization of canine osteosarcoma by array comparative genomic hybridization and RT-qPCR: Signatures of genomic imbalance in canine osteosarcoma parallel the human counterpart. Genes Chromosomes Cancer 2011, 50, 859–874. [Google Scholar] [CrossRef]
- Fossey, S.L.; Liao, A.T.; McCleese, J.K.; Bear, M.D.; Lin, J.; Li, P.-K.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 activation and expression in canine and human osteosarcoma. BMC Cancer 2009, 9, 81. [Google Scholar] [CrossRef]
- Gardner, H.L.; Sivaprakasam, K.; Briones, N.; Zismann, V.; Perdigones, N.; Drenner, K.; Facista, S.; Richholt, R.; Liang, W.; Aldrich, J.; et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun. Biol. 2019, 2, 266. [Google Scholar] [CrossRef] [PubMed]
- Marley, K.; Bracha, S.; Seguin, B. Osteoprotegerin activates osteosarcoma cells that co-express RANK and RANKL. Exp. Cell Res. 2015, 338, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.; Davis, S.; Lana, S.; Withrow, S.; Sangiorgi, L.; Picci, P.; Hewitt, S.; Triche, T.; Meltzer, P.; Khanna, C. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genom. 2009, 10, 625. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef]
- Flint, A.F.; U’Ren, L.; Legare, M.E.; Withrow, S.J.; Dernell, W.; Hanneman, W.H. Overexpression of the erbB-2 Proto-oncogene in Canine Osteosarcoma Cell Lines and Tumors. Vet. Pathol. 2004, 41, 291–296. [Google Scholar] [CrossRef]
- Shahabi, V.; Seavey, M.M.; Maciag, P.C.; Rivera, S.; Wallecha, A. Development of a live and highly attenuated Listeria monocytogenes-based vaccine for the treatment of Her2/neu-overexpressing cancers in human. Cancer Gene Ther. 2011, 18, 53–62. [Google Scholar] [CrossRef]
- Musser, M.L.; Berger, E.P.; Tripp, C.D.; Clifford, C.A.; Bergman, P.J.; Johannes, C.M. Safety evaluation of the canine osteosarcoma vaccine, live Listeria vector. Vet. Comp. Oncol. 2021, 19, 92–98. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Ma, W.; Xue, R.; Zhu, Z.; Farrukh, H.; Song, W.; Li, T.; Zheng, L.; Pan, C.-x. Increasing cure rates of solid tumors by immune checkpoint inhibitors. Exp. Hematol. Oncol. 2023, 12, 10. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Ikebuchi, R.; Okagawa, T.; Adachi, M.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; Murata, S.; et al. Expression of PD-L1 on canine tumor cells and enhancement of IFN-γ production from tumor-infiltrating cells by PD-L1 blockade. PLoS ONE 2014, 9, e98415. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, V.B.; Perry, S.N.; Todd, M.; Huckle, W.R.; LeRoith, T. PD-1, PD-L1, and PD-L2 Gene Expression and Tumor Infiltrating Lymphocytes in Canine Melanoma. Vet. Pathol. 2021, 58, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Coy, J.; Caldwell, A.; Chow, L.; Guth, A.; Dow, S. PD-1 expression by canine T cells and functional effects of PD-1 blockade. Vet. Comp. Oncol. 2017, 15, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.; Elmslie, R.; Dow, S.; Guth, A. Checkpoint molecule expression by B and T cell lymphomas in dogs. Vet. Comp. Oncol. 2018, 16, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Withers, S.S.; Chang, H.; Spanier, J.A.; De La Trinidad, V.L.; Panesar, H.; Fife, B.T.; Sciammas, R.; Sparger, E.E.; Moore, P.F.; et al. Development of canine PD-1/PD-L1 specific monoclonal antibodies and amplification of canine T cell function. PLoS ONE 2020, 15, e0235518. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, Y.; Shosu, K.; Okuda, M.; Noguchi, S.; Mizuno, T. Development and characterization of monoclonal antibodies against canine PD-1 and PD-L1. Vet. Immunol. Immunopathol. 2018, 198, 19–25. [Google Scholar] [CrossRef]
- Hartley, G.; Faulhaber, E.; Caldwell, A.; Coy, J.; Kurihara, J.; Guth, A.; Regan, D.; Dow, S. Immune regulation of canine tumour and macrophage PD-L1 expression. Vet. Comp. Oncol. 2017, 15, 534–549. [Google Scholar] [CrossRef]
- Mason, N.J.; Chester, N.; Xiong, A.; Rotolo, A.; Wu, Y.; Yoshimoto, S.; Glassman, P.; Gulendran, G.; Siegel, D.L. Development of a fully canine anti-canine CTLA4 monoclonal antibody for comparative translational research in dogs with spontaneous tumors. mAbs 2021, 13, 2004638. [Google Scholar] [CrossRef]
- Pantelyushin, S.; Ranninger, E.; Guerrera, D.; Hutter, G.; Maake, C.; Markkanen, E.; Bettschart-Wolfensberger, R.; Rohrer Bley, C.; Läubli, H.; vom Berg, J. Cross-Reactivity and Functionality of Approved Human Immune Checkpoint Blockers in Dogs. Cancers 2021, 13, 785. [Google Scholar] [CrossRef]
- Nascimento, C.; Urbano, A.C.; Gameiro, A.; Ferreira, J.; Correia, J.; Ferreira, F. Serum PD-1/PD-L1 Levels, Tumor Expression and PD-L1 Somatic Mutations in HER2-Positive and Triple Negative Normal-Like Feline Mammary Carcinoma Subtypes. Cancers 2020, 12, 1386. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.S.; Stickings, P.; White, J.M.; Neal, S.; Crowcroft, N.S.; Sesardic, D.; Efstratiou, A. A review of the international issues surrounding the availability and demand for diphtheria antitoxin for therapeutic use. Vaccine 2009, 28, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, A.M. Clemens Freiherr von Pirquet: Explaining immune complex disease in 1906. Nat. Immunol. 2000, 1, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L. Ten years of Orthoclone OKT3 (muromonab-CD3): A review. J. Transpl. Coord. Off. Publ. N. Am. Transpl. Coord. Organ. 1996, 6, 109–119, quiz 120–121. [Google Scholar] [CrossRef]
- Sgro, C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: Bibliographic review. Toxicology 1995, 105, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kimball, J.A.; Norman, D.J.; Shield, C.F.; Schroeder, T.J.; Lisi, P.; Garovoy, M.; O’Connell, J.B.; Stuart, F.; McDiarmid, S.V.; Wall, W. The OKT3 antibody response study: A multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ tranplantation. Transpl. Immunol. 1995, 3, 212–221. [Google Scholar] [CrossRef]
- Ducourau, E.; Mulleman, D.; Paintaud, G.; Miow Lin, D.C.; Lauféron, F.; Ternant, D.; Watier, H.; Goupille, P. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res. Ther. 2011, 13, R105. [Google Scholar] [CrossRef]
- van den Bemt, B.J.F.; den Broeder, A.A.; Wolbink, G.J.; Hekster, Y.A.; van Riel, P.L.C.M.; Benraad, B.; van den Hoogen, F.H.J. Anti-infliximab antibodies are already detectable in most patients with rheumatoid arthritis halfway through an infusioncycle: An open-label pharmacokinetic cohort study. BMC Musculoskelet. Disord. 2011, 12, 12. [Google Scholar] [CrossRef]
- Baert, F.; Noman, M.; Vermeire, S.; Van Assche, G.; D’Haens, G.; Carbonez, A.; Rutgeerts, P. Influence of Immunogenicity on the Long-Term Efficacy of Infliximab in Crohn’s Disease. N. Engl. J. Med. 2003, 348, 601–608. [Google Scholar] [CrossRef]
- Haraoui, B.; Cameron, L.; Ouellet, M.; White, B. Anti-infliximab antibodies in patients with rheumatoid arthritis who require higher doses of infliximab to achieve or maintain a clinical response. J. Rheumatol. 2006, 33, 31–36. [Google Scholar]
- Bou-Assaly, W.; Mukherji, S. Cetuximab (erbitux). AJNR. Am. J. Neuroradiol. 2010, 31, 626–627. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, B.H.; Allen, R.; Spigel, D.R.; Stinchcombe, T.E.; Moore, D.T.; Berlin, J.D.; Goldberg, R.M. High Incidence of Cetuximab-Related Infusion Reactions in Tennessee and North Carolina and the Association with Atopic History. J. Clin. Oncol. 2007, 25, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef]
- Carson, A.S.; Gardner, A.; Iweala, O.I. Where’s the Beef? Understanding Allergic Responses to Red Meat in Alpha-Gal Syndrome. J. Immunol. 2022, 208, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment with Monoclonal Antibodies. Front. Immunol. 2020, 11, 01951. [Google Scholar] [CrossRef]
- Jeglum, K.A. Chemoimmunotherapy of Canine Lymphoma with Adjuvant Canine Monoclonal Antibody 231. Vet. Clin. N. Am. Small Anim. Pract. 1996, 26, 73–85. [Google Scholar] [CrossRef]
- Beirão, B.C.B.; Raposo, T.; Jain, S.; Hupp, T.; Argyle, D.J. Challenges and opportunities for monoclonal antibody therapy in veterinary oncology. Vet. J. 2016, 218, 40–50. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef]
- Michels, G.M.; Walsh, K.F.; Kryda, K.A.; Mahabir, S.P.; Walters, R.R.; Hoevers, J.D.; Martinon, O.M. A blinded, randomized, placebo-controlled trial of the safety of lokivetmab (ZTS-00103289), a caninized anti-canine IL-31 monoclonal antibody in client-owned dogs with atopic dermatitis. Vet. Dermatol. 2016, 27, 505-e136. [Google Scholar] [CrossRef]
- Van Brussel, L.; Moyaert, H.; Escalada, M.; Mahabir, S.P.; Stegemann, M.R. A masked, randomised clinical trial evaluating the efficacy and safety of lokivetmab compared to saline control in client-owned dogs with allergic dermatitis. Vet. Dermatol. 2021, 32, 477-e131. [Google Scholar] [CrossRef]
- Souza, C.P.; Rosychuk, R.A.W.; Contreras, E.T.; Schissler, J.R.; Simpson, A.C. A retrospective analysis of the use of lokivetmab in the management of allergic pruritus in a referral population of 135 dogs in the western USA. Vet. Dermatol. 2018, 29, 489-e164. [Google Scholar] [CrossRef] [PubMed]
- Gearing, D.P.; Huebner, M.; Virtue, E.R.; Knight, K.; Hansen, P.; Lascelles, B.D.X.; Gearing, R.P.; Drew, A.C. In Vitro and In Vivo Characterization of a Fully Felinized Therapeutic Anti-Nerve Growth Factor Monoclonal Antibody for the Treatment of Pain in Cats. J. Vet. Intern. Med. 2016, 30, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Lascelles, B.D.X.; Knazovicky, D.; Case, B.; Freire, M.; Innes, J.F.; Drew, A.C.; Gearing, D.P. A canine-specific anti-nerve growth factor antibody alleviates pain and improves mobility and function in dogs with degenerative joint disease-associated pain. BMC Vet. Res. 2015, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Gearing, D.P.; Virtue, E.R.; Gearing, R.P.; Drew, A.C. A fully caninised anti-NGF monoclonal antibody for pain relief in dogs. BMC Vet. Res. 2013, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Igase, M.; Nemoto, Y.; Itamoto, K.; Tani, K.; Nakaichi, M.; Sakurai, M.; Sakai, Y.; Noguchi, S.; Kato, M.; Tsukui, T.; et al. A pilot clinical study of the therapeutic antibody against canine PD-1 for advanced spontaneous cancers in dogs. Sci. Rep. 2020, 10, 18311. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Takagi, S.; Kagawa, Y.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Deguchi, T.; Nakajima, C.; et al. A canine chimeric monoclonal antibody targeting PD-L1 and its clinical efficacy in canine oral malignant melanoma or undifferentiated sarcoma. Sci. Rep. 2017, 7, 8951. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Asano, Y.; Sajiki, Y.; Deguchi, T.; Okagawa, T.; Watari, K.; Takeuchi, H.; Takagi, S.; Hosoya, K.; et al. Exploration of serum biomarkers in dogs with malignant melanoma receiving anti-PD-L1 therapy and potential of COX-2 inhibition for combination therapy. Sci. Rep. 2022, 12, 9265. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Nishimura, M.; Kagawa, Y.; Takagi, S.; Hosoya, K.; Ohta, H.; Kim, S.; Okagawa, T.; Izumi, Y.; et al. PD-L1 immunohistochemistry for canine cancers and clinical benefit of anti-PD-L1 antibody in dogs with pulmonary metastatic oral malignant melanoma. npj Precis. Oncol. 2021, 5, 10. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995, 374, 168–173. [Google Scholar] [CrossRef]
- Bathula, N.V.; Bommadevara, H.; Hayes, J.M. Nanobodies: The Future of Antibody-Based Immune Therapeutics. Cancer Biother. Radiopharm. 2021, 36, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Applications of Nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Saga, T.; Neumann, R.D.; Heya, T.; Sato, J.; Kinuya, S.; Le, N.; Paik, C.H.; Weinstein, J.N. Targeting cancer micrometastases with monoclonal antibodies: A binding-site barrier. Proc. Natl. Acad. Sci. USA 1995, 92, 8999–9003. [Google Scholar] [CrossRef] [PubMed]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of Monoclonal Antibodies in Solid Tumors: Direct Experimental Evidence for a Binding Site Barrier. Cancer Res. 1992, 52, 5144–5153. [Google Scholar] [PubMed]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biologics 2019, 13, 33–51. [Google Scholar] [CrossRef]
- Thurber, G.M.; Zajic, S.C.; Wittrup, K.D. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. J. Nucl. Med. 2007, 48, 995–999. [Google Scholar] [CrossRef]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef]
- Debie, P.; Lafont, C.; Defrise, M.; Hansen, I.; van Willigen, D.M.; van Leeuwen, F.W.B.; Gijsbers, R.; D’Huyvetter, M.; Devoogdt, N.; Lahoutte, T.; et al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J. Control. Release 2020, 317, 34–42. [Google Scholar] [CrossRef]
- Bannas, P.; Lenz, A.; Kunick, V.; Well, L.; Fumey, W.; Rissiek, B.; Haag, F.; Schmid, J.; Schütze, K.; Eichhoff, A.; et al. Molecular imaging of tumors with nanobodies and antibodies: Timing and dosage are crucial factors for improved in vivo detection. Contrast Media Mol. Imaging 2015, 10, 367–378. [Google Scholar] [CrossRef]
- Beltrán Hernández, I.; Rompen, R.; Rossin, R.; Xenaki, K.T.; Katrukha, E.A.; Nicolay, K.; van Bergen en Henegouwen, P.; Grüll, H.; Oliveira, S. Imaging of Tumor Spheroids, Dual-Isotope SPECT, and Autoradiographic Analysis to Assess the Tumor Uptake and Distribution of Different Nanobodies. Mol. Imaging Biol. 2019, 21, 1079–1088. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Yang, Y.; Balthasar, J.P. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021, 81, 4145–4154. [Google Scholar] [CrossRef] [PubMed]
- Rossotti, M.A.; Bélanger, K.; Henry, K.A.; Tanha, J. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2021, 289, 4304–4327. [Google Scholar] [CrossRef] [PubMed]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.C.; Wurthner, J.U.; Morley, P.J.; Birchler, M.A.; Lambert, J.; Albayaty, M.; Serone, A.P.; Wilson, R.; Chen, Y.; Forrest, R.M.; et al. Autoantibodies to Variable Heavy (VH) Chain Ig Sequences in Humans Impact the Safety and Clinical Pharmacology of a VH Domain Antibody Antagonist of TNF-α Receptor 1. J. Clin. Immunol. 2013, 33, 1192–1203. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody® targeting the DR5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Promises and Challenges as Lifesaving Treatments. Int. J. Mol. Sci. 2022, 23, 5009. [Google Scholar] [CrossRef]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Wang, J.; Kang, G.; Yuan, H.; Cao, X.; Huang, H.; de Marco, A. Research Progress and Applications of Multivalent, Multispecific and Modified Nanobodies for Disease Treatment. Front. Immunol. 2022, 12, 838082. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Bartunek, J.; Barbato, E.; Heyndrickx, G.; Vanderheyden, M.; Wijns, W.; Holz, J.-B. Novel Antiplatelet Agents: ALX-0081, a Nanobody Directed towards von Willebrand Factor. J. Cardiovasc. Transl. Res. 2013, 6, 355–363. [Google Scholar] [CrossRef]
- Ishiwatari-Ogata, C.; Kyuuma, M.; Ogata, H.; Yamakawa, M.; Iwata, K.; Ochi, M.; Hori, M.; Miyata, N.; Fujii, Y. Ozoralizumab, a Humanized Anti-TNFα NANOBODY® Compound, Exhibits Efficacy Not Only at the Onset of Arthritis in a Human TNF Transgenic Mouse but Also during Secondary Failure of Administration of an Anti-TNFα IgG. Front. Immunol. 2022, 13, 853008. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Weinberg, M.A.; Morris, A.; Reich, K. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: A multicentre, randomised, placebo-controlled, phase 2b study. Lancet 2021, 397, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; ‘t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Zhao, Z.; Li, Y.; Mustafa, B.; Chen, Z.; Barve, A.; Jain, A.; Yao, X.; Li, G.; et al. Discovery of Anti-PD-L1 Human Domain Antibodies for Cancer Immunotherapy. Front. Immunol. 2022, 13, 838966. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, H.; Wang, X.; Bai, Y.; Wang, P.; Wu, J.; Jiang, X.; Wang, Y.; Cai, H.; Xu, T.; et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discov. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Broos, K.; Lecocq, Q.; Xavier, C.; Bridoux, J.; Nguyen, T.T.; Corthals, J.; Schoonooghe, S.; Lion, E.; Raes, G.; Keyaerts, M.; et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers 2019, 11, 872. [Google Scholar] [CrossRef]
- Sun, Z.; Li, W.; Mellors, J.W.; Orentas, R.; Dimitrov, D.S. Construction of a Large Size Human Immunoglobulin Heavy Chain Variable (VH) Domain Library, Isolation and Characterization of Novel Human Antibody VH Domains Targeting PD-L1 and CD22. Front. Immunol. 2022, 13, 869825. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Harb, W.; Peer, C.J.; Hua, Q.; Xu, S.; Lu, H.; Lu, N.; He, Y.; Xu, T.; Dong, R.; et al. First-in-Human Phase I Study of Envafolimab, a Novel Subcutaneous Single-Domain Anti-PD-L1 Antibody, in Patients with Advanced Solid Tumors. Oncologist 2021, 26, e1514–e1525. [Google Scholar] [CrossRef]
- Li, J.; Deng, Y.; Zhang, W.; Zhou, A.-P.; Guo, W.; Yang, J.; Yuan, Y.; Zhu, L.; Qin, S.; Xiang, S.; et al. Subcutaneous envafolimab monotherapy in patients with advanced defective mismatch repair/microsatellite instability high solid tumors. J. Hematol. Oncol. 2021, 14, 95. [Google Scholar] [CrossRef]
- Breen, M.; Modiano, J.F. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans–man and his best friend share more than companionship. Chromosome Res. 2008, 16, 145–154. [Google Scholar] [CrossRef]
- Ito, D.; Frantz, A.M.; Modiano, J.F. Canine lymphoma as a comparative model for human non-Hodgkin lymphoma: Recent progress and applications. Vet. Immunol. Immunopathol. 2014, 159, 192–201. [Google Scholar] [CrossRef]
- Ponce, F.; Marchal, T.; Magnol, J.P.; Turinelli, V.; Ledieu, D.; Bonnefont, C.; Pastor, M.; Delignette, M.L.; Fournel-Fleury, C. A morphological study of 608 cases of canine malignant lymphoma in France with a focus on comparative similarities between canine and human lymphoma morphology. Vet. Pathol. 2010, 47, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.A.; Xu, T.; Goodall, C.; Rhodes, A.C.; Kashyap, A.; He, J.; Bracha, S. Cross-species analysis of the canine and human bladder cancer transcriptome and exome. Genes Chromosomes Cancer 2017, 56, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, D.; Hahn, N.M.; Ramos-Vara, J.A.; Knapp, D.W. Naturally-occurring canine invasive urothelial carcinoma harbors luminal and basal transcriptional subtypes found in human muscle invasive bladder cancer. PLoS Genet. 2018, 14, e1007571. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.W.; Dhawan, D.; Ramos-Vara, J.A.; Ratliff, T.L.; Cresswell, G.M.; Utturkar, S.; Sommer, B.C.; Fulkerson, C.M.; Hahn, N.M. Naturally-Occurring Invasive Urothelial Carcinoma in Dogs, a Unique Model to Drive Advances in Managing Muscle Invasive Bladder Cancer in Humans. Front. Oncol. 2020, 9, 01493. [Google Scholar] [CrossRef]
- Knapp, D.W.; Ramos-Vara, J.A.; Moore, G.E.; Dhawan, D.; Bonney, P.L.; Young, K.E. Urinary Bladder Cancer in Dogs, a Naturally Occurring Model for Cancer Biology and Drug Development. ILAR J. 2014, 55, 100–118. [Google Scholar] [CrossRef]
- Caliari, D.; Zappulli, V.; Rasotto, R.; Cardazzo, B.; Frassineti, F.; Goldschmidt, M.H.; Castagnaro, M. Triple-negative vimentin-positive heterogeneous feline mammary carcinomas as a potential comparative model for breast cancer. BMC Vet. Res. 2014, 10, 185. [Google Scholar] [CrossRef]
- De Maria, R.; Olivero, M.; Iussich, S.; Nakaichi, M.; Murata, T.; Biolatti, B.; Di Renzo, M.F. Spontaneous Feline Mammary Carcinoma Is a Model of HER2 Overexpressing Poor Prognosis Human Breast Cancer. Cancer Res. 2005, 65, 907–912. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).