


Changes in Gut Microbiota in Peruvian Cattle Genetic Nucleus by Breed and Correlations with Beef Quality
 , , , ,
, , , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiment and Sample Collection
2.2. Meat Quality Traits Detection
2.3. Analyses of Blood Parameters
2.4. DNA Extraction and Sequencing
2.5. Bioinformatics Analysis
2.6. Statistics Analysis
3. Results
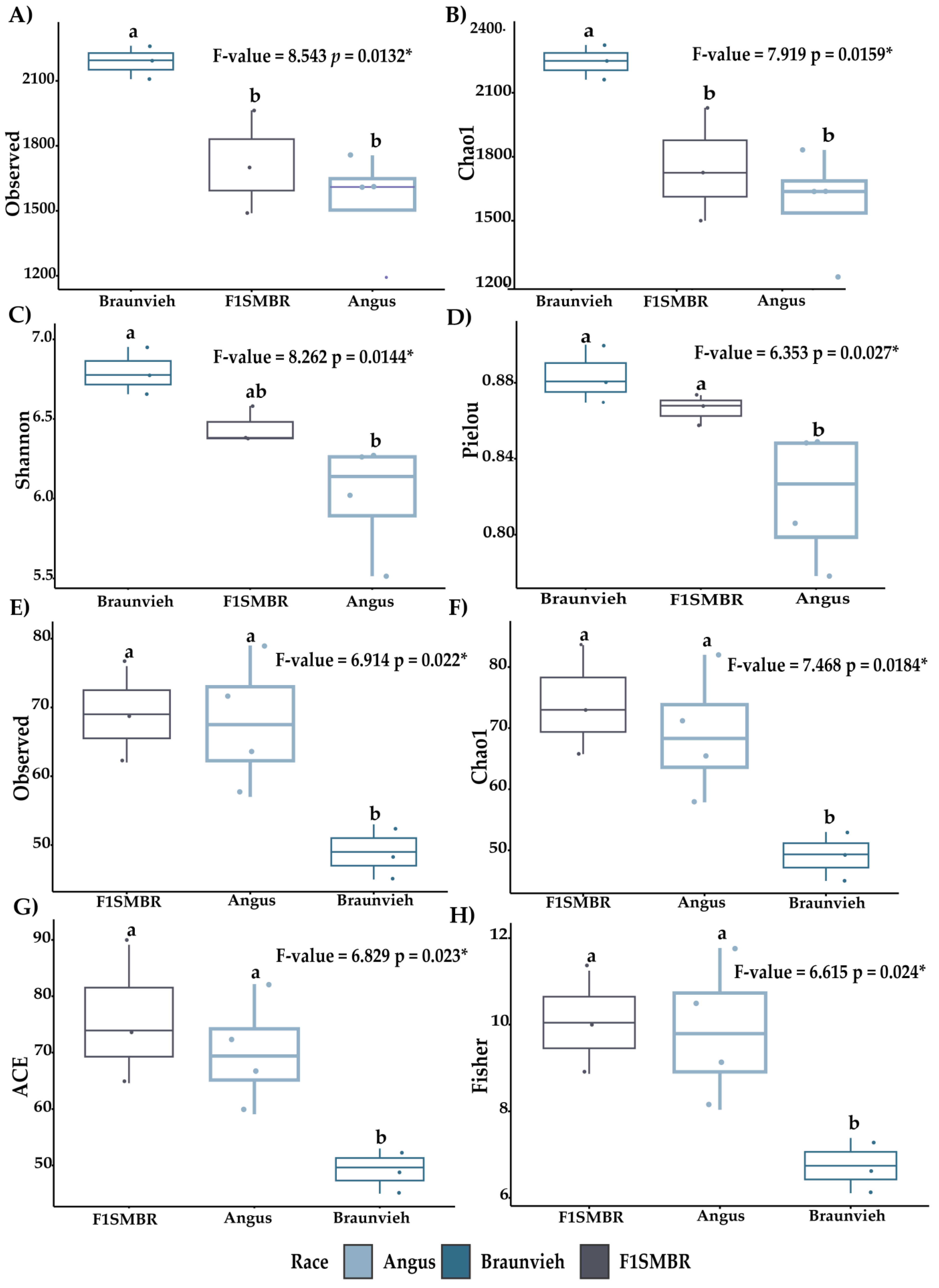
3.1. Analysis of Effect of Breed on the Gut Microbiota Diversity and Composition
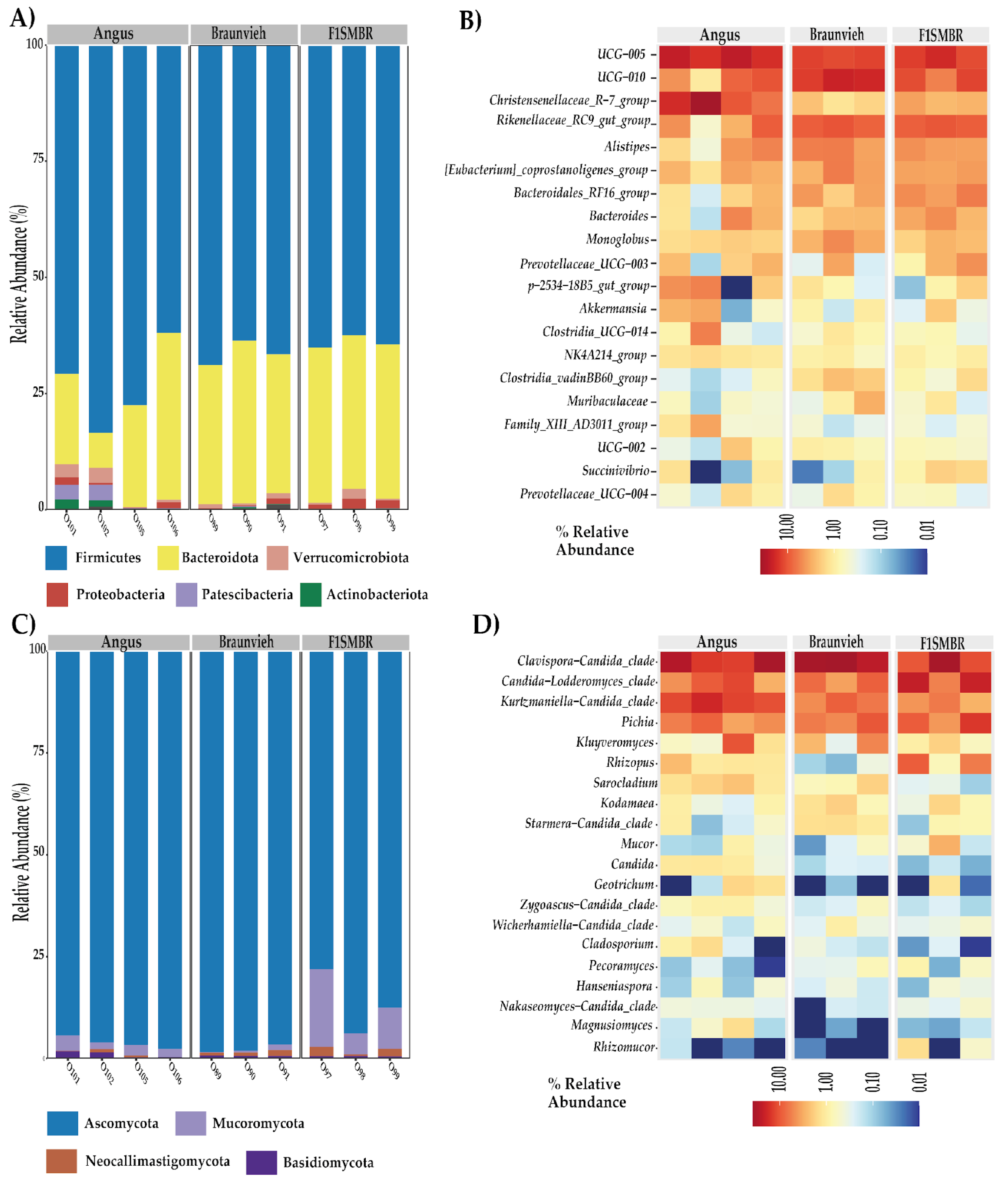
3.2. Effect of Breed on the Gut Microbiota Taxonomy
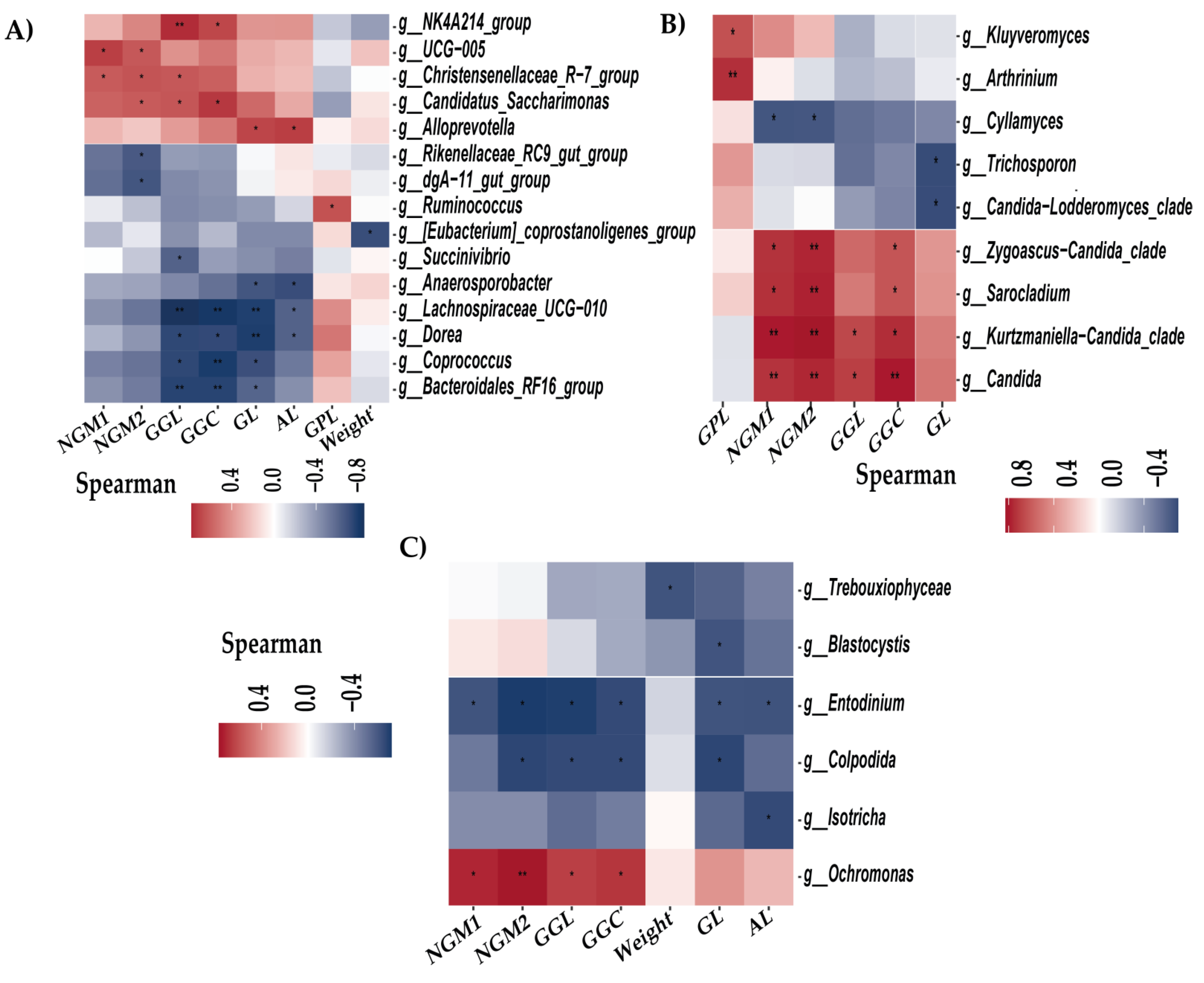
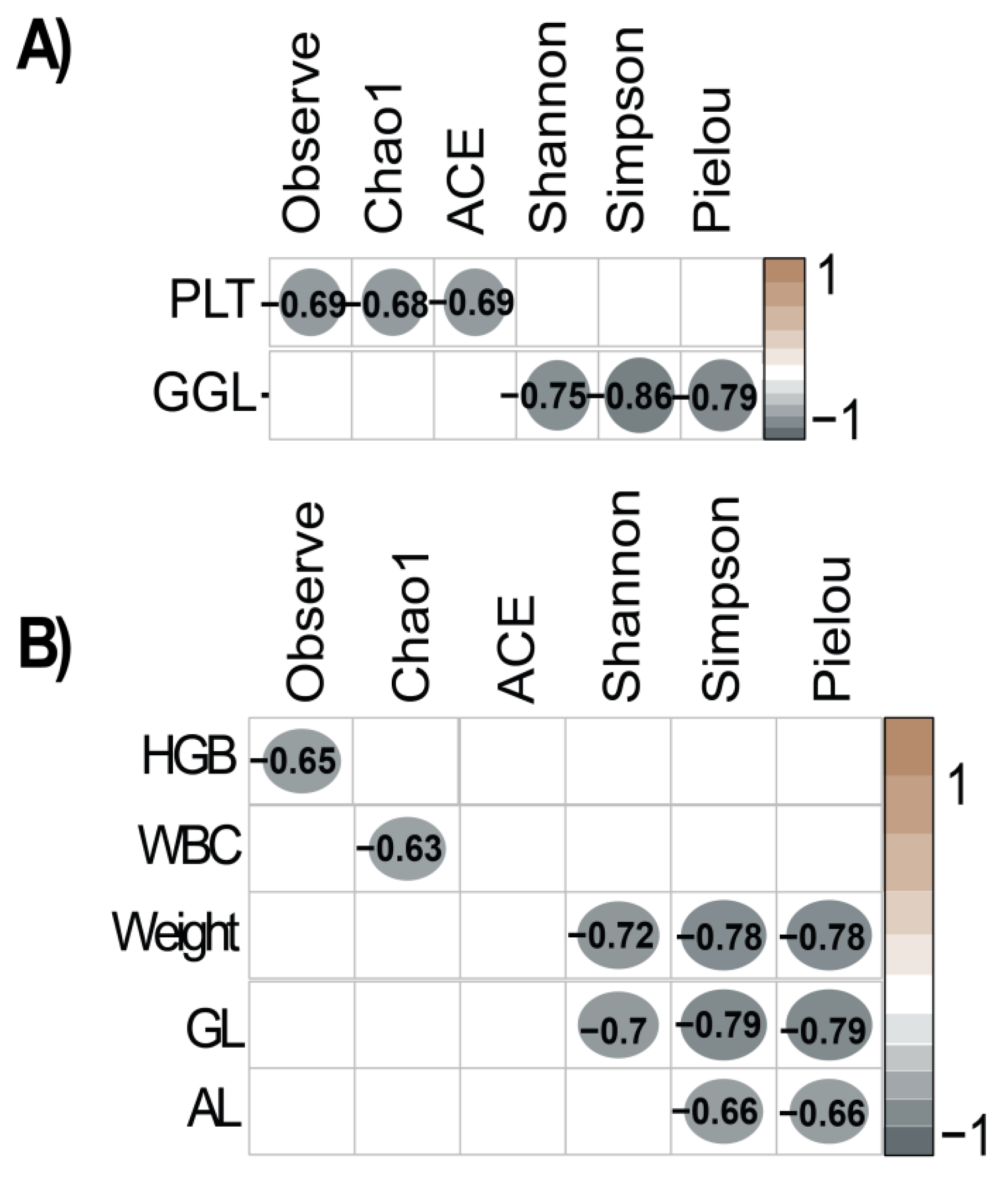
3.3. Relationship Between Gut Microbiota and Beef Quality Variables
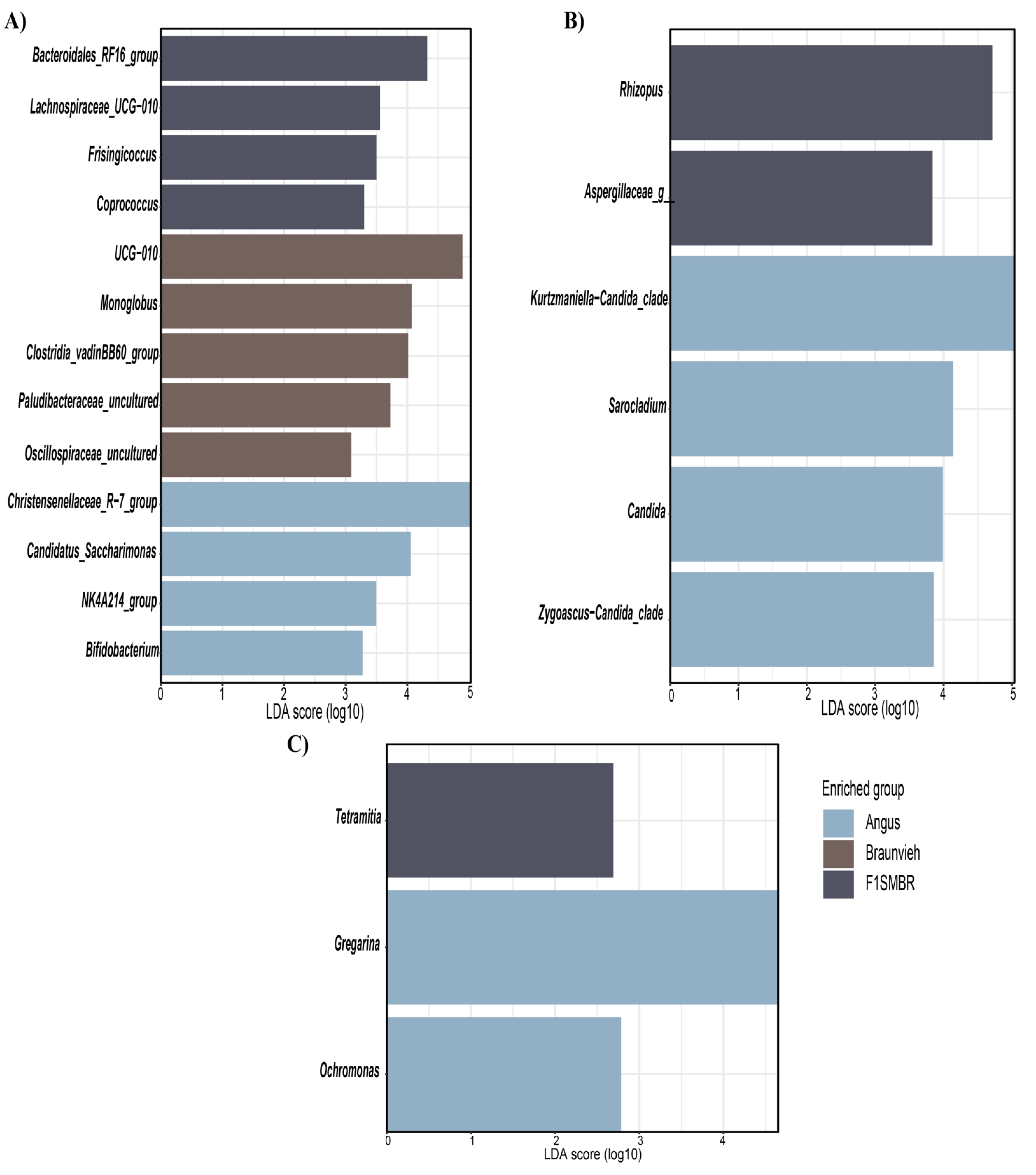
3.4. Biomarkers Identification for Different Breed
3.5. Breed Relationship of Alpha/Beta Diversity with Variables
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Wang, H.; Lin, L.; Miao, C.; Zhang, Y.; Zhou, B. Intestinal Barrier Damage Involved in Intestinal Microflora Changes in Fluoride-Induced Mice. Chemosphere 2019, 234, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, B.; Li, F.; He, Y.; Wang, L.; Fakhar-e-Alam Kulyar, M.; Li, H.; Fu, Y.; Zhu, H.; Wang, Y.; et al. Integrated Bacterial and Fungal Diversity Analysis Reveals the Gut Microbial Alterations in Diarrheic Giraffes. Front. Microbiol. 2021, 12, 712092. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, D.; Zhang, Y.; Li, K.; Wang, M.; Ma, J. Dynamic Distribution of Gut Microbiota in Cattle at Different Breeds and Health States. Front. Microbiol. 2023, 14, 1113730. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host Genetics Influence the Rumen Microbiota and Heritable Rumen Microbial Features Associate with Feed Efficiency in Cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef]
- Abbas, W.; Howard, J.T.; Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Erickson, G.E.; Spangler, M.L.; Fernando, S.C. Influence of Host Genetics in Shaping the Rumen Bacterial Community in Beef Cattle. Sci. Rep. 2020, 10, 15101. [Google Scholar] [CrossRef]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A Heritable Subset of the Core Rumen Microbiome Dictates Dairy Cow Productivity and Emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef]
- Sasson, G.; Kruger Ben-Shabat, S.; Seroussi, E.; Doron-Faigenboim, A.; Shterzer, N.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Halperin, E.; Mizrahi, I. Heritable Bovine Rumen Bacteria Are Phylogenetically Related and Correlated with the Cow’s Capacity to Harvest Energy from Its Feed. mBio 2017, 8, e00703-17. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You Are What You Eat: Diet, Health and the Gut Microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Canibe, N.; O’Dea, M.; Abraham, S. Potential Relevance of Pig Gut Content Transplantation for Production and Research. J. Anim. Sci. Biotechnol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-Species Comparisons of Host Genetic Associations with the Microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, K.; Xiang, Y.; Zhou, W.; Gui, G.; Yang, H. The Fecal Microbiota Composition of Boar Duroc, Yorkshire, Landrace and Hampshire Pigs. Asian-Australas. J. Anim. Sci. 2017, 30, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Pandit, R.J.; Hinsu, A.T.; Patel, N.V.; Koringa, P.G.; Jakhesara, S.J.; Thakkar, J.R.; Shah, T.M.; Limon, G.; Psifidi, A.; Guitian, J.; et al. Microbial Diversity and Community Composition of Caecal Microbiota in Commercial and Indigenous Indian Chickens Determined Using 16s rDNA Amplicon Sequencing. Microbiome 2018, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, Y.; Liang, J.; Wu, Y.; Wright, A.; Liao, X. Exploratory Analysis of the Microbiological Potential for Efficient Utilization of Fiber Between Lantang and Duroc Pigs. Front. Microbiol. 2018, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wright, A.-D.G.; Si, H.; Wang, X.; Qian, W.; Zhang, Z.; Li, G. Changes in the Rumen Microbiome and Metabolites Reveal the Effect of Host Genetics on Hybrid Crosses. Environ. Microbiol. Rep. 2016, 8, 1016–1023. [Google Scholar] [CrossRef]
- Bainbridge, M.L.; Cersosimo, L.M.; Wright, A.-D.G.; Kraft, J. Rumen Bacterial Communities Shift across a Lactation in Holstein, Jersey and Holstein × Jersey Dairy Cows and Correlate to Rumen Function, Bacterial Fatty Acid Composition and Production Parameters. FEMS Microbiol. Ecol. 2016, 92, fiw059. [Google Scholar] [CrossRef]
- Hernandez-Sanabria, E.; Goonewardene, L.A.; Wang, Z.; Zhou, M.; Moore, S.S.; Guan, L.L. Influence of Sire Breed on the Interplay among Rumen Microbial Populations Inhabiting the Rumen Liquid of the Progeny in Beef Cattle. PLoS ONE 2013, 8, e58461. [Google Scholar] [CrossRef]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef]
- Gagaoua, M.; Picard, B. Current Advances in Meat Nutritional, Sensory and Physical Quality Improvement. Foods 2020, 9, 321. [Google Scholar] [CrossRef]
- Sakowski, T.; Grodkowski, G.; Gołebiewski, M.; Slósarz, J.; Kostusiak, P.; Solarczyk, P.; Puppel, K. Genetic and Environmental Determinants of Beef Quality—A Review. Front. Vet. Sci. 2022, 9, 819605. [Google Scholar] [CrossRef]
- Petracci, M.; Soglia, F.; Berri, C. Chapter 3—Muscle Metabolism and Meat Quality Abnormalities. In Poultry Quality Evaluation; Petracci, M., Berri, C., Eds.; Woodhead Publishing Series in Food Science, Technology and Nutrition; Woodhead Publishing: Sawston, UK, 2017; pp. 51–75. ISBN 978-0-08-100763-1. [Google Scholar]
- Gerber, P.J.; Mottet, A.; Opio, C.I.; Falcucci, A.; Teillard, F. Environmental Impacts of Beef Production: Review of Challenges and Perspectives for Durability. Meat Sci. 2015, 109, 2–12. [Google Scholar] [CrossRef]
- Thorslund, C.A.H.; Sandøe, P.; Aaslyng, M.D.; Lassen, J. A Good Taste in the Meat, a Good Taste in the Mouth—Animal Welfare as an Aspect of Pork Quality in Three European Countries. Livest. Sci. 2016, 193, 58–65. [Google Scholar] [CrossRef]
- Joo, S.T.; Kim, G.D.; Hwang, Y.H.; Ryu, Y.C. Control of Fresh Meat Quality through Manipulation of Muscle Fiber Characteristics. Meat Sci. 2013, 95, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Corredor, F.-A.; Figueroa, D.; Estrada, R.; Salazar, W.; Quilcate, C.; Vásquez, H.V.; Gonzales, J.; Maicelo, J.L.; Medina, P.; Arbizu, C.I. Genetic Diversity and Population Structure of a Peruvian Cattle Herd Using SNP Data. Front. Genet. 2023, 14, 1073843. [Google Scholar] [CrossRef] [PubMed]
- Marín, G.Y.G.; Torre-Trinidad, L.L.; Farje-Muguruza, P.; Vargas, J.F.G. Desarrollo y calidad embrionaria de un protocolo de superovulación en vacas Brahman en el distrito de Codo del Pozuzo, Huánuco, Perú. Braz. J. Anim. Environ. Res. 2024, 7, e70052. [Google Scholar] [CrossRef]
- Chen, D.; Wang, X.; Guo, Q.; Deng, H.; Luo, J.; Yi, K.; Sun, A.; Chen, K.; Shen, Q. Muscle Fatty Acids, Meat Flavor Compounds and Sensory Characteristics of Xiangxi Yellow Cattle in Comparison to Aberdeen Angus. Animals 2022, 12, 1161. [Google Scholar] [CrossRef]
- Conanec, A.; Campo, M.; Richardson, I.; Ertbjerg, P.; Failla, S.; Panea, B.; Chavent, M.; Saracco, J.; Williams, J.L.; Ellies-Oury, M.-P.; et al. Has Breed Any Effect on Beef Sensory Quality? Livest. Sci. 2021, 250, 104548. [Google Scholar] [CrossRef]
- Cziszter, L.-T.; Ilie, D.-E.; Neamt, R.-I.; Neciu, F.-C.; Saplacan, S.-I.; Gavojdian, D. Comparative Study on Production, Reproduction and Functional Traits between Fleckvieh and Braunvieh Cattle. Asian-Australas. J. Anim. Sci. 2017, 30, 666–671. [Google Scholar] [CrossRef]
- Burrow, H.M. Genetic Aspects of Cattle Adaptation in the Tropics. Genet. Cattle 2015, 571–597. [Google Scholar] [CrossRef]
- Anderson, C.J.; Koester, L.R.; Schmitz-Esser, S. Rumen Epithelial Communities Share a Core Bacterial Microbiota: A Meta-Analysis of 16S rRNA Gene Illumina MiSeq Sequencing Datasets. Front. Microbiol. 2021, 12, 625400. [Google Scholar] [CrossRef]
- Vaulot, D.; Geisen, S.; Mahé, F.; Bass, D. Pr2-Primers: An 18S rRNA Primer Database for Protists. Mol. Ecol. Resour. 2022, 22, 168–179. [Google Scholar] [CrossRef]
- Mishra, P.; Tulsani, N.J.; Jakhesara, S.J.; Dafale, N.A.; Patil, N.V.; Purohit, H.J.; Koringa, P.G.; Joshi, C.G. Exploring the Eukaryotic Diversity in Rumen of Indian Camel (Camelus Dromedarius) Using 18S rRNA Amplicon Sequencing. Arch. Microbiol. 2020, 202, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.S.; Mikhailov, I.S.; Petrova, D.P.; Galachyants, Y.P.; Zakharova, Y.R.; Likhoshway, Y.V. The Effect of Metabarcoding 18S rRNA Region Choice on Diversity of Microeukaryotes Including Phytoplankton. World J. Microbiol. Biotechnol. 2023, 39, 229. [Google Scholar] [CrossRef] [PubMed]
- Treven, P.; Mahnič, A.; Rupnik, M.; Golob, M.; Pirš, T.; Matijašić, B.B.; Lorbeg, P.M. Evaluation of Human Milk Microbiota by 16S rRNA Gene Next-Generation Sequencing (NGS) and Cultivation/MALDI-TOF Mass Spectrometry Identification. Front. Microbiol. 2019, 10, 2612. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Dueholm, M.S.; McIlroy, S.J.; Kirkegaard, R.H.; Nielsen, P.H.; Albertsen, M. Retrieval of a Million High-Quality, Full-Length Microbial 16S and 18S rRNA Gene Sequences without Primer Bias. Nat. Biotechnol. 2018, 36, 190–195. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- R Core Team. R A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R Package for Data Mining in Microbial Community Ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Xu, S.; Zhan, L.; Tang, W.; Wang, Q.; Dai, Z.; Zhou, L.; Feng, T.; Chen, M.; Wu, T.; Hu, E.; et al. MicrobiotaProcess: A Comprehensive R Package for Deep Mining Microbiome. Innovation 2023, 4, 100388. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, P.; Zhou, X.; Fu, M.; Wang, T.; Liu, Z.; Liu, X.; Wang, Z.; Liu, B. Effect of Host Genetics and Gut Microbiome on Fat Deposition Traits in Pigs. Front. Microbiol. 2022, 13, 925200. [Google Scholar] [CrossRef]
- Liu, S.; Cao, R.; Liu, L.; Lv, Y.; Qi, X.; Yuan, Z.; Fan, X.; Yu, C.; Guan, Q. Correlation Between Gut Microbiota and Testosterone in Male Patients with Type 2 Diabetes Mellitus. Front. Endocrinol. 2022, 13, 836485. [Google Scholar] [CrossRef]
- Motta, G.A.; Neto, P.S.M.; Nociti, R.P.; Santana, Á.E. Hematological Normality, Serum Biochemistry, and Acute Phase Proteins in Healthy Beef Calves in the Brazilian Savannah. Animals 2023, 13, 2398. [Google Scholar] [CrossRef] [PubMed]
- Geletu, U.S.; Usmael, M.A.; Mummed, Y.Y.; Ibrahim, A.M. Quality of Cattle Meat and Its Compositional Constituents. Vet. Med. Int. 2021, 2021, 7340495. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Nelson, C.D.; Driver, J.D.; Elzo, M.A.; Jeong, K.C. Animal Breed Composition Is Associated with the Hindgut Microbiota Structure and β-Lactam Resistance in the Multibreed Angus-Brahman Herd. Front. Microbiol. 2019, 10, 1846. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.-B.; Meng, J.-X.; Ma, H.; Liu, R.; Qin, Y.; Qin, Y.-F.; Geng, H.-L.; Ni, H.-B.; Zhang, X.-X. Description of Gut Mycobiota Composition and Diversity of Caprinae Animals. Microbiol. Spectr. 2023, 11, e0242422. [Google Scholar] [CrossRef]
- Parfrey, L.W.; Walters, W.A.; Lauber, C.L.; Clemente, J.C.; Berg-Lyons, D.; Teiling, C.; Kodira, C.; Mohiuddin, M.; Brunelle, J.; Driscoll, M.; et al. Communities of Microbial Eukaryotes in the Mammalian Gut within the Context of Environmental Eukaryotic Diversity. Front. Microbiol. 2014, 5, 298. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Chen, Y.; Wu, H.; Meng, Q.; Guan, L.L. Metatranscriptomic Profiling Reveals the Effect of Breed on Active Rumen Eukaryotic Composition in Beef Cattle with Varied Feed Efficiency. Front. Microbiol. 2020, 11, 367. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Prenafeta-Boldú, F.; Zingaretti, L.M.; Gonzalez-Rodriguez, O.; Dalmau, A.; Quintanilla, R.; Ballester, M. Gut Eukaryotic Communities in Pigs: Diversity, Composition and Host Genetics Contribution. Anim. Microbiome 2020, 2, 18. [Google Scholar] [CrossRef]
- Chen, Z.; Sun, Y.; Chen, L.; Zhang, Y.; Wang, J.; Li, H.; Yan, X.; Xia, L.; Yao, G. Differences in Meat Quality between Angus Cattle and Xinjiang Brown Cattle in Association with Gut Microbiota and Its Lipid Metabolism. Front. Microbiol. 2022, 13, 988984. [Google Scholar] [CrossRef]
- Daghio, M.; Ciucci, F.; Buccioni, A.; Cappucci, A.; Casarosa, L.; Serra, A.; Conte, G.; Viti, C.; McAmmond, B.M.; Van Hamme, J.D.; et al. Correlation of Breed, Growth Performance, and Rumen Microbiota in Two Rustic Cattle Breeds Reared Under Different Conditions. Front. Microbiol. 2021, 12, 652031. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Zhu, L.; Xu, Y.; Liu, N.; Sun, X.; Hu, L.; Huang, H.; Wei, K.; Zhu, R. Dynamic Distribution of Gut Microbiota in Goats at Different Ages and Health States. Front. Microbiol. 2018, 9, 2509. [Google Scholar] [CrossRef]
- Zapata, C.; Estrada, R.; Oros, O.; Sánchez, D.; Maicelo, J.L.; Arbizu, C.I.; Coila, P. Alterations in the Gut Microbial Composition and Diversity Associated with Diarrhea in Neonatal Peruvian Alpacas. Small Rumin. Res. 2024, 235, 107273. [Google Scholar] [CrossRef]
- Carroll, C.; Olsen, K.D.; Ricks, N.J.; Dill-McFarland, K.A.; Suen, G.; Robinson, T.F.; Chaston, J.M. Bacterial Communities in the Alpaca Gastrointestinal Tract Vary with Diet and Body Site. Front. Microbiol. 2019, 9, 3334. [Google Scholar] [CrossRef] [PubMed]
- Estrada, R.; Romero, Y.; Figueroa, D.; Coila, P.; Hañari-Quispe, R.D.; Aliaga, M.; Galindo, W.; Alvarado, W.; Casanova, D.; Quilcate, C. Effects of Age in Fecal Microbiota and Correlations with Blood Parameters in Genetic Nucleus of Cattle. Microorganisms 2024, 12, 1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, P.; Liu, X.; Zhang, C.; Lu, Q.; Xi, D.; Yang, R.; Wang, S.; Bai, W.; Yang, Z.; et al. The Composition of Fungal Communities in the Rumen of Gayals (Bos Frontalis), Yaks (Bos Grunniens), and Yunnan and Tibetan Yellow Cattle (Bos Taurs). Pol. J. Microbiol. 2019, 68, 505–514. [Google Scholar] [CrossRef]
- Zhao, G.; Niu, Y.; Wang, H.; Qin, S.; Zhang, R.; Wu, Y.; Xiao, X.; Xu, Y.; Yang, C. Effects of Three Different Plant-Derived Polysaccharides on Growth Performance, Immunity, Antioxidant Function, and Cecal Microbiota of Broilers. J. Sci. Food Agric. 2024, 104, 1020–1029. [Google Scholar] [CrossRef]
- Nan, S.; Li, J.; Kuang, Y.; Feng, J.; Wang, H.; Niu, J.; Wu, Y.; Zhang, W.; Nie, C. Differential Microbial Composition and Interkingdom Interactions in the Intestinal Microbiota of Holstein and German Simmental × Holstein Cross F1 Calves: A Comprehensive Analysis of Bacterial and Fungal Diversity. Microorganisms 2024, 12, 486. [Google Scholar] [CrossRef]
- Zhu, Y.; Cidan, Y.; Sun, G.; Li, X.; Shahid, M.A.; Luosang, Z.; Suolang, Z.; Suo, L.; Basang, W. Comparative Analysis of Gut Fungal Composition and Structure of the Yaks under Different Feeding Models. Front. Vet. Sci. 2023, 10, 1193558. [Google Scholar] [CrossRef]
- Mann, A.E.; Mazel, F.; Lemay, M.A.; Morien, E.; Billy, V.; Kowalewski, M.; Di Fiore, A.; Link, A.; Goldberg, T.L.; Tecot, S.; et al. Biodiversity of Protists and Nematodes in the Wild Nonhuman Primate Gut. ISME J. 2020, 14, 609–622. [Google Scholar] [CrossRef]
- Geng, X.; Liu, Y.; Xu, W.; Li, G.; Xue, B.; Feng, Y.; Tang, S.; Wei, W.; Yuan, H. Eukaryotes May Play an Important Ecological Role in the Gut Microbiome of Graves’ Disease. Front. Immunol. 2024, 15, 1334158. [Google Scholar] [CrossRef]
- Dubik, M.; Pilecki, B.; Moeller, J.B. Commensal Intestinal Protozoa—Underestimated Members of the Gut Microbial Community. Biology 2022, 11, 1742. [Google Scholar] [CrossRef]
- Yang, C.; Tsedan, G.; Liu, Y.; Hou, F. Shrub Coverage Alters the Rumen Bacterial Community of Yaks (Bos Grunniens) Grazing in Alpine Meadows. J. Anim. Sci. Technol. 2020, 62, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, B.; Wang, Y.; Bai, J.; Gao, Y.; Ru, X.; Bi, C.; Li, J.; Shan, A. Effects of Sex on Fat Deposition through Gut Microbiota and Short-Chain Fatty Acids in Weaned Pigs. Anim. Nutr. 2024, 17, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of Different Partial 16S rRNA Gene Sequence Regions for Phylogenetic Analysis of Microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique Aspects of Fiber Degradation by the Ruminal Ethanologen Ruminococcus Albus 7 Revealed by Physiological and Transcriptomic Analysis. BMC Genom. 2014, 15, 1066. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Nourrisson, C.; Scanzi, J.; Brunet, J.; Delbac, F.; Dapoigny, M.; Poirier, P. Prokaryotic and Eukaryotic Fecal Microbiota in Irritable Bowel Syndrome Patients and Healthy Individuals Colonized with Blastocystis. Front. Microbiol. 2021, 12, 713347. [Google Scholar] [CrossRef]
- Rajamanikam, A.; Isa, M.N.M.; Samudi, C.; Devaraj, S.; Govind, S.K. Gut Bacteria Influence Blastocystis Sp. Phenotypes and May Trigger Pathogenicity. PLoS Negl. Trop. Dis. 2023, 17, e0011170. [Google Scholar] [CrossRef]
- Sanchez, D.; Zapata, C.; Romero, Y.; Flores-Huarco, N.H.; Oros, O.; Alvarado, W.; Quilcate, C.; Guevara-Alvarado, H.M.; Estrada, R.; Coila, P. Parasitism-Induced Changes in Microbial Eukaryotes of Peruvian Alpaca Gastrointestinal Tract. Life 2024, 14, 187. [Google Scholar] [CrossRef]
- Herrera, G.; Vega, L.; Patarroyo, M.A.; Ramírez, J.D.; Muñoz, M. Gut Microbiota Composition in Health-Care Facility-and Community-Onset Diarrheic Patients with Clostridioides Difficile Infection. Sci. Rep. 2021, 11, 10849. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, J.; Wang, X.; Han, L.; Yang, Y.; Wang, Q.; Yu, Q. Metagenomic and Transcriptomic Analyses Reveal the Differences and Associations Between the Gut Microbiome and Muscular Genes in Angus and Chinese Simmental Cattle. Front. Microbiol. 2022, 13, 815915. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; An, M.; Zhang, W.; Li, K.; Kulyar, M.F.-E.-A.; Duan, K.; Zhou, H.; Wu, Y.; Wan, X.; Li, J.; et al. Integrated Bacteria-Fungi Diversity Analysis Reveals the Gut Microbial Changes in Buffalo with Mastitis. Front. Vet. Sci. 2022, 9, 918541. [Google Scholar] [CrossRef] [PubMed]
- Eldesouky, I.; Mohamed, N.; Khalaf, D.; Salama, A.; Elsify, A.; Ombarak, R.; El-Ballal, S.; Effat, M.; Shabrawy, M.A.L. Candida Mastitis in Dairy Cattle with Molecular Detection of Candida Albicans.|EBSCOhost. Available online: https://openurl.ebsco.com/contentitem/doi:10.9775%2Fkvfd.2015.14843?sid=ebsco:plink:crawler&id=ebsco:doi:10.9775%2Fkvfd.2015.14843 (accessed on 6 November 2024).
- Ji, Y.; Dong, X.; Liu, Z.; Wang, W.; Yan, H.; Liu, X. Effects of Bovine Pichia Kudriavzevii T7, Candida Glabrata B14, and Lactobacillus Plantarum Y9 on Milk Production, Quality and Digestive Tract Microbiome in Dairy Cows. Microorganisms 2022, 10, 842. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.; Luo, H.; Wang, Y.; Liu, X.; Zhou, X. Epidemiological Investigation of Non-Albicans Candida Species Recovered from Mycotic Mastitis of Cows in Yinchuan, Ningxia of China. BMC Vet. Res. 2018, 14, 251. [Google Scholar] [CrossRef]
- Wang, M.; Wan, J.; Rong, H.; He, F.; Wang, H.; Zhou, J.; Cai, C.; Wang, Y.; Xu, R.; Yin, Z.; et al. Alterations in Gut Glutamate Metabolism Associated with Changes in Gut Microbiota Composition in Children with Autism Spectrum Disorder. mSystems 2019, 4, e00321-18. [Google Scholar] [CrossRef]
- Park, T.; Yu, Z. Interactions between Entodinium Caudatum and an Amino Acid-Fermenting Bacterial Consortium: Fermentation Characteristics and Protozoal Population in Vitro. J. Anim. Sci. Technol. 2023, 65, 387. [Google Scholar] [CrossRef]
- Holman, D.B.; Gzyl, K.E.; Scott, H.; Prieto, N.; López-Campos, Ó. Cara Service Associations between the Rumen Microbiota and Carcass Merit and Meat Quality in Beef Cattle. Appl. Microbiol. Biotechnol. 2024, 108, 287. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, C.; Huo, D.; Hu, Q.; Peng, Q. Comparative Study of the Gut Microbiome Potentially Related to Milk Protein in Murrah Buffaloes (Bubalus Bubalis) and Chinese Holstein Cattle. Sci. Rep. 2017, 7, 42189. [Google Scholar] [CrossRef]
- Sim, S.; Lee, H.; Yoon, S.; Seon, H.; Park, C.; Kim, M. The Impact of Different Diets and Genders on Fecal Microbiota in Hanwoo Cattle. J. Anim. Sci. Technol. 2022, 64, 897. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Wu, C. Modulation of Gut Microbiota and Immune System by Probiotics, Pre-Biotics, and Post-Biotics. Front. Nutr. 2022, 8, 634897. [Google Scholar] [CrossRef]
- Zhao, G.; Xiang, Y.; Wang, X.; Dai, B.; Zhang, X.; Ma, L.; Yang, H.; Lyu, W. Exploring the Possible Link between the Gut Microbiome and Fat Deposition in Pigs. Oxid. Med. Cell. Longev. 2022, 2022, 1098892. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Stensvold, C.R.; Jirků-Pomajbíková, K.; Parfrey, L.W. Are Human Intestinal Eukaryotes Beneficial or Commensals? PLoS Pathog. 2015, 11, e1005039. [Google Scholar] [CrossRef] [PubMed]
- Laforest-Lapointe, I.; Arrieta, M.-C. Microbial Eukaryotes: A Missing Link in Gut Microbiome Studies. mSystems 2018, 3, e00201-17. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, Y.; Guan, L.L. Translational Gut Microbiome Research for Strategies to Improve Beef Cattle Production Sustainability and Meat Quality. Anim. Biosci. 2024, 37, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Teng, J.; Wang, X.; Xu, B.; Niu, Y.; Ma, L.; Yan, X. Multi-Omics Analysis Reveals Gut Microbiota-Induced Intramuscular Fat Deposition via Regulating Expression of Lipogenesis-Associated Genes. Anim. Nutr. 2022, 9, 84–99. [Google Scholar] [CrossRef]
- Marestone, B.S.; Torres Junior, R.A.A.; Silva, L.O.C.; Menezes, G.R.O.; Muniz, C.A.S.D.; Ribeiro, E.L.A. Genetic Parameters for Traditional and Novel Ultrasound Carcass Traits in Nellore Cattle. Trop. Anim. Health Prod. 2022, 54, 34. [Google Scholar] [CrossRef]
- Naserkheil, M.; Lee, D.-H.; Kong, H.-S.; Seong, J.; Mehrban, H. Estimation of Genetic Parameters and Correlation between Yearling Ultrasound Measurements and Carcass Traits in Hanwoo Cattle. Animals 2021, 11, 1425. [Google Scholar] [CrossRef]
- Miller, G.A.; Hyslop, J.J.; Barclay, D.; Edwards, A.; Thomson, W.; Duthie, C.-A. Using 3D Imaging and Machine Learning to Predict Liveweight and Carcass Characteristics of Live Finishing Beef Cattle. Front. Sustain. Food Syst. 2019, 3, 30. [Google Scholar] [CrossRef]
- Ferdous, T.; Jiang, L.; Dinu, I.; Groizeleau, J.; Kozyrskyj, A.L.; Greenwood, C.M.T.; Arrieta, M.-C. The Rise to Power of the Microbiome: Power and Sample Size Calculation for Microbiome Studies. Mucosal Immunol. 2022, 15, 1060–1070. [Google Scholar] [CrossRef]
- Kers, J.G.; Saccenti, E. The Power of Microbiome Studies: Some Considerations on Which Alpha and Beta Metrics to Use and How to Report Results. Front. Microbiol. 2022, 12, 796025. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feed Type | Amount | Dry Matter (%) | Moisture (%) | Crude Protein (%) | Metabolizable Energy (Mcal/kg DM) | Crude Fiber (%) | Calcium (%) | Phosphorus (%) |
|---|---|---|---|---|---|---|---|---|
| Corn Silage | 10% of body weight | 24–26 | 74–76 | 7.85 | 2.25 | 27 | 0.29 | 0.17 |
| Balanced Feed | 2 kg | 90 | 10 | 14.21 | 2.88 | 4.75 | 0.31 | 0.65 |
| Balanced Feed | % |
|---|---|
| Ground yellow corn | 58.14 |
| Soybean meal | 14.53 |
| Wheat bran | 23.26 |
| Baking soda | 1.57 |
| Common salt | 0.5 |
| Mineral salts | 2 |
| Total | 100 |
| Race | GPL | GGL | GL | AL | GPC | GGC | NGM1 | NGM2 |
|---|---|---|---|---|---|---|---|---|
| Angus | 6.31 | 4.12 | 6.35 | 55.14 | 4.00 | 2.83 | 162.20 | 131.16 |
| Braunvieh | 6.72 | 3.32 | 6.58 | 61.13 | 4.51 | 2.23 | 107.19 | 91.71 |
| F1(SMxBR) | 6.14 | 3.18 | 5.34 | 46.35 | 3.56 | 1.24 | 83.52 | 71.16 |
| Bray-Curtis | Jaccard | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | ||||||||||
| Df | SumOfSqs | R2 | F | p-value | Df | SumOfSqs | R2 | F | p-value | |
| Race | 2 | 0.61835 | 0.3339 | 2.0051 | 0.001 *** | 2 | 0.8116 | 0.28707 | 1.6106 | 0.001 *** |
| Residual | 8 | 1.23357 | 0.6661 | 8 | 2.0156 | 0.71293 | 0.0003 *** | |||
| Total | 10 | 1.85192 | 1 | 10 | 2.8272 | 1 | ||||
| Fungi | ||||||||||
| Df | SumOfSqs | R2 | F | p-value | Df | SumOfSqs | R2 | F | p-value | |
| Race | 2 | 0.25335 | 0.3723 | 2.3724 | 0.0053 ** | 2 | 0.48255 | 0.33902 | 2.0516 | 0.0044 ** |
| Residual | 8 | 0.42715 | 0.6277 | 8 | 0.94084 | 0.66098 | ||||
| Total | 10 | 0.6805 | 1 | 10 | 1.42339 | 1 | ||||
| Protists | ||||||||||
| Df | SumOfSqs | R2 | F | p-value | Df | SumOfSqs | R2 | F | p-value | |
| Race | 2 | 0.5995 | 0.34142 | 2.0737 | 0.019 * | 2 | 0.79217 | 0.30164 | 1.7277 | 0.0156 * |
| Residual | 8 | 1.11869 | 0.65858 | 8 | 1.83401 | 0.69836 | ||||
| Total | 10 | 1.69864 | 1 | 10 | 2.62618 | 1 | ||||
| Bacteria | Bray | Jaccard | ||||||
|---|---|---|---|---|---|---|---|---|
| Mantel Test | Partial Mantel Test | Mantel Test | Partial Mantel Test | |||||
| Variables | r | p | r | p | r | p | r | p |
| GGL | 0.409 | 0.019 | 0.408 | 0.017 | 0.308 | 0.028 | 0.31 | 0.037 |
| NMG1 | 0.407 | 0.007 | 0.347 | 0.02 | 0.328 | 0.026 | 0.417 | 0.003 |
| NGM2 | 0.376 | 0.019 | 0.385 | 0.016 | ||||
| Fungi | ||||||||
| Variables | r | p | r | p | r | p | r | p |
| RBC | 0.524 | 0.01 | 0.467 | 0.009 | ||||
| MCV | 0.649 | 0.012 | 0.612 | 0.005 | ||||
| MCHC | 0.697 | 0.008 | 0.672 | 0.004 | 0.343 | 0.008 | ||
| NGM1 | 0.307 | 0.035 | 0.341 | 0.024 | ||||
| NGM2 | 0.315 | 0.02 | 0.281 | 0.03 | ||||
| GGC | 0.373 | 0.011 | 0.38 | 0.007 | ||||
| GGL | ||||||||
| Protist | ||||||||
| Variables | r | p | r | p | r | p | r | p |
| NEU | 0.297 | 0.044 | 0.293 | 0.031 | ||||
| SEG | 0.297 | 0.043 | 0.293 | 0.045 | ||||
| NGM1 | 0.434 | 0.001 | 0.422 | 0.004 | ||||
| NGM2 | 0.495 | 0.001 | 0.487 | 0.002 | ||||
| PLT | 0.388 | 0.025 | 0.379 | 0.036 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quilcate, C.; Estrada, R.; Romero, Y.; Rojas, D.; Mamani, R.; Hañari-Quispe, R.D.; Aliaga, M.; Galindo, W.; Vásquez, H.V.; Maicelo, J.L.; et al. Changes in Gut Microbiota in Peruvian Cattle Genetic Nucleus by Breed and Correlations with Beef Quality. Vet. Sci. 2024, 11, 608. https://doi.org/10.3390/vetsci11120608
Quilcate C, Estrada R, Romero Y, Rojas D, Mamani R, Hañari-Quispe RD, Aliaga M, Galindo W, Vásquez HV, Maicelo JL, et al. Changes in Gut Microbiota in Peruvian Cattle Genetic Nucleus by Breed and Correlations with Beef Quality. Veterinary Sciences. 2024; 11(12):608. https://doi.org/10.3390/vetsci11120608
Chicago/Turabian StyleQuilcate, Carlos, Richard Estrada, Yolanda Romero, Diorman Rojas, Rolando Mamani, Renán Dilton Hañari-Quispe, Mery Aliaga, Walter Galindo, Héctor V. Vásquez, Jorge L. Maicelo, and et al. 2024. "Changes in Gut Microbiota in Peruvian Cattle Genetic Nucleus by Breed and Correlations with Beef Quality" Veterinary Sciences 11, no. 12: 608. https://doi.org/10.3390/vetsci11120608
APA StyleQuilcate, C., Estrada, R., Romero, Y., Rojas, D., Mamani, R., Hañari-Quispe, R. D., Aliaga, M., Galindo, W., Vásquez, H. V., Maicelo, J. L., & Arbizu, C. I. (2024). Changes in Gut Microbiota in Peruvian Cattle Genetic Nucleus by Breed and Correlations with Beef Quality. Veterinary Sciences, 11(12), 608. https://doi.org/10.3390/vetsci11120608