Simple Summary
Infectious coryza is an acute respiratory infectious disease of chickens caused by Av. paragallinarum. Av. paragallinarum can be serotyped into A (A1–A4), B (B-1), and C (C1–C4) serovars. However, there is no effective molecular typing scheme to gain basic knowledge about the Av. paragallinarum population structure. MLST has played a major role in investigating the genetic structure of bacterial populations and has rapidly become the cornerstone technique for the molecular typing of pathogenic microorganisms. In this study, we are the first to establish this method and applied to PubMLST for a public MLST site for Av. paragallinarum. More samples and the ST sequence data can help to clearly observe the genetic evolution and epidemic patterns of Av. paragallinarum.
Abstract
Infectious coryza is an acute respiratory infection caused by Avibacterium paragallinarum, which is widely distributed throughout the world. However, there is no effective molecular typing scheme to obtain basic knowledge about the Av. paragallinarum population structure. This study aimed to develop a multilocus sequence typing (MLST) scheme for Av. paragallinarum that allows for the worldwide comparison of sequence data. For this purpose, the genetic variability of 59 Av. paragallinarum strains from different geographical origins and serovars was analyzed to identify correlations. The MLST scheme was developed using seven conserved housekeeping genes, which identified eight STs that clustered all of the strains into three evolutionary branches. The analytical evaluation of the clone group relationship between the STs revealed two clone complexes (CC1 and CC2) and three singletons (ST2, ST5, and ST6). Most of the isolates from China belonged to ST1 and ST3 in CC1. ST8 from Peru and ST7 from North America together formed CC2. Our results showed that the Av. paragallinarum strains isolated from China had a distant genetic relationship with CC2, indicating strong regional specificity. The MLST scheme established in this study can monitor the dynamics and genetic differences of Av. paragallinarum transmission.
1. Introduction
Avibacterium paragallinarum, a member of the Pasteurellaceae family, is a Gram-negative bacterium responsible for infectious coryza (IC) in chickens, causing significant economic losses to the poultry industry [1]. The clinical course of IC typically includes severe respiratory tract infection with nasal discharge, facial swelling, sneezing, and infraorbital sinus swelling, resulting in growth retardation and reduced egg production [1]. In general, the symptoms persist for two to three weeks [2]. Asymptomatic infected birds and carriers play a crucial role in the spread of the disease [2,3]. Chickens serve as natural hosts for Av. paragallinarum. There are two typing schemes for Av. paragallinarum: the Page scheme and the Kume scheme. The Page scheme divides Av. paragallinarum into three serovars, A, B, and C, based on the plate agglutination assay [4]. Kume classified Av. paragallinarum into three distinct groups based on the hemagglutination inhibition (HI) test [5]. Blackall et al. regrouped and renamed the Kume protocol to match the Page serovars of A, B, and C, and subdivided it into nine serovars, A-1 to A-4, B-1, and C-1 to C-4 [6]. The Page scheme remains the most commonly used serotyping scheme for Av. paragallinarum, as inactivated vaccines only protect against the Page serogroup present in the vaccine [7,8,9]. The Page scheme requires three serovar-specific antibodies, whereas the Kume scheme requires nine serovar-specific antibodies, all of which are complicated and require chicken erythrocytes fixed with glutaraldehyde and antigens treated with potassium thiocyanate or hyaluronidase [10]. Consequently, only a handful of laboratories that specialize in relevant research can achieve the serovar classification of Av. paragallinarum [11]. In addition, some isolates fail to display HA activity even after undergoing hyaluronidase treatment and, therefore, cannot be serotyped by the HI test [12].
IC occurs globally, including in China, Japan, South Korea, India, Bangladesh, and Thailand in Asia; the United Kingdom and the Netherlands in Europe; South Africa in Africa; Mexico and the United States in North America; and Peru in South America. The serovars A, B, and C of Av. paragallinarum strains are widely distributed, and the epidemic strains differ between countries. In China, serovar A was first reported in 1987, serovar C in 1993, and serovar B in 2003 [13]. Before 2000, most of the isolated strains were of serovar A. Since 2000, serovar C strains were isolated gradually. Between 2012 and 2017, serovar B was the most common type isolated in China [13,14]. In recent years, serovars A and C have become more prevalent. The incidence of IC has risen in China, and the disease has even occurred in chickens immunized with inactivated vaccines, indicating that the inactivated vaccine has insufficient immune protection against the current epidemic strains [15]. A previous study showed that the variant strains and increased virulence lead to decreased inactivated vaccine protection [15]. It is difficult to compare strains around the world and exchange information across institutes. To effectively control the spread of Av. paragallinarum, it is essential to implement rapid, accurate, and convenient strain typing methods to accurately determine its evolutionary trajectory.
Sakamoto et al. proposed a multiplex PCR and PCR-RFLP method based on the hypervariable region of the HMTp210 gene for the serotyping of Av. paragallinarum [16]. However, it has been demonstrated that these methods are not suitable for differentiating the serovar of Av. paragallinarum isolates [11]. Soriano et al. claimed that ERIC-PCR could be used as a laboratory molecular typing tool for Av. paragallinarum [17]. Subsequently, this method was also evaluated, and the data indicated that ERIC-PCR is not ideal for the molecular typing of Av. paragallinarum serovars [18]. Multilocus sequence analysis (MLSA) of Avibacterium was conducted to classify Pasteurellaceae. The authors highlight the advantages of MLSA over 16S rRNA sequence analysis. It was found that Av. paragallinarum is a distinct species, while Av. volantium, Av. avium, Av. gallinarum, Av. endocarditis, and Avibacterium sp. A form a complex of species that are difficult to distinguish. MLSA is suitable for the initial typing and phylogenetic analysis of new populations [19]. Another scheme, multilocus sequence typing (MLST), is suitable for the fine typing of known populations. MLST is an ideal choice for long-term epidemiological monitoring and global strain comparison due to its standardization and ease of data sharing. There is currently no mature molecular typing method for Av. paragallinarum.
MLST is a method that utilizes the well-established principles of multilocus enzyme electrophoresis [20], which detects amino acid sequence changes in enzymes associated with the activity of basic cell families [21]. MLST determines the genetic information of each strain through the internal fragments of seven housekeeping genes, each about 500 bp nucleotides in length. The different sequences of each gene are referred to as alleles, and the combination of seven alleles creates an allelic map that defines the sequence type (ST). The housekeeping genes are highly conserved, making it a slow process for mutations to occur. As a result, MLST offers a reliable and precise method for evaluating isolates with high resolution. This technique has proven to be an invaluable tool for analyzing genetic relationships between strains and can be applied to study the phylogeny and molecular epidemiology of pathogens [22].
MLST allows researchers to compare data from different laboratories around the world for the same pathogenic microorganism quickly and efficiently [23]. This method has played a major role in investigating the genetic structure of bacterial populations and rapidly became the cornerstone technique for molecular typing of pathogenic microorganisms [24]. Since the establishment and application of MLST in Neisseria meningitidis in 1998 [25], MLST has been applied to a wide range of bacteria and has become the gold standard for bacterial molecular typing [26]. However, it has not yet been implemented in Av. paragallinarum. In this study, we developed an MLST typing method for Av. paragallinarum based on strains isolated in recent years and whole-genome data from the GenBank. We applied to PubMLST for a public MLST site of Av. paragallinarum for reference and use by other researchers.
2. Materials and Methods
2.1. Bacterial Isolates and Strains
A total of 59 strains of Av. paragallinarum were used to develop the MLST, including 44 field strains, 2 reference strains, and 13 complete genome sequences (Table 1). Av. paragallinarum strains (29 strains) were isolated from the chickens with clinical symptoms of IC and conserved in our laboratory [27]; 14 strains were isolated from the infraorbital sinuses of chickens with clinical symptoms of IC in this study. Additionally, strain 2021/06 was sent to us by a biological company in June 2021 for serotyping. Most of the field strains were isolated from Jiangsu, while a few were isolated from Ningxia, Hebei, and Hubei. Reference strains 221 and H-18 were purchased from the China Veterinary Culture Collection Center (CVCC). The complete genome sequences of 13 strains were obtained from GenBank. The details of all 59 Av. paragallinarum strains are shown in Table 1.

Table 1.
Background information of 59 Av. paragallinarum strains.
Av. paragallinarum isolates were cultured on trypticase soy agar (Hopebio Biotech Co., Ltd., Qingdao, China) with 5% fetal bovine serum (Lonsera Biotech Co., Ltd., Suzhou, China) and 0.0025% reduced nicotinamide adenine dinucleotide (Sangon Biotech Co., Ltd., Shanghai, China) at 37 °C in 5% CO2 for 24–48 h. The colony morphology was translucent with a diameter of approximately 2 mm. The specific Hpg-2 primers (F: TGAGGGTAGTCTTGCACGCGAAT; R: CAAGGTATCGATCGTCTCTCTACT) were used to identify Av. paragallinarum [28]. The serovar of the isolates was determined by the HI test [12]. The antisera for the HI test were obtained from our previous study [27]. The isolates’ culture was centrifuged and treated with potassium thiocyanate (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) at 4 °C for 2 h. Then, the isolates were sonicated and washed with PBS as antigens. The chicken erythrocytes were fixed with glutaraldehyde (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China). The serovar of the isolate corresponds to the antiserum with the highest HI titer.
2.2. Selection of Housekeeping Genes for MLST
Based on the MLST analysis of Pasteurellaceae obtained from the PubMLST database (https://pubmlst.org/organisms, accessed on 27 September 2022) [29], 14 housekeeping genes were selected from the genomic sequence of Av. paragallinarum. The primers were designed to target conserved regions within these sequences, and partial segments within the genes were delineated using Primer5 software (Version 5.0). Based on the determination principles of housekeeping genes, 7 genes were ultimately identified by comparing the genomic sequence of Av. paragallinarum. The primers are listed in Table 2. The positional distribution of the housekeeping genes on the chromosome was as follows: pmi, 248.0 kb; infB, 632.4 kb; mdh, 708.1 kb; adk, 1316.9 kb; deoD, 1792.6 kb; recA, 2432.3 kb; zwf, 2489.2 kb. The minimum distance between the housekeeping genes was 56.9 kb.
Table 2.
Primer sequences of 7 housekeeping genes.
2.3. Gene Amplification and Sequencing
PCR was performed in a total volume of 25 μL containing 2.5 μL of 10× EasyTaq® buffer, 2 μL of 2.5 mM dNTPs, 0.5 μL of forward and reverse primers (10 μM), 0.25μL of EasyTaq® DNA Polymerase (Transgen Biotech Co., Ltd., Beijing, China), and 1 μL of genomic DNA templates. The thermocycling parameters were as follows: initial denaturation at 94 °C for 5 min, followed by 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s for 35 cycles, completed with a final extension at 72 °C for 7 min. The PCR products were sent to General Biol Co., Ltd. (Anhui, China) for sequencing. The obtained sequences were edited for further analysis.
2.4. MLST Analysis
For each housekeeping gene, the nucleotide sequences of 59 strains were compared, and the different sequences were assigned allele numbers (different alleles were numbered in ascending order). The Av. paragallinarum strains were characterized by distinct allelic combinations of 7 loci, which defined the allelic profile. The distinct allelic profiles were designated STs. The number of polymorphic sites and the number of alleles were calculated using DnaSP software (Version 5.10.0001). The average GC content was calculated using DNASTAR software (Version 7.1.0.44). Pairwise ratios of nonsynonymous substitutions to synonymous substitutions (dN/dS) were calculated by the method of modified Nei–Gojobori with the calculated R of the MEGA X software (Version 10.2.6) [30]. The neutral theory of molecular evolution test for housekeeping genes was performed using the statistical method Tajima’s D in DnaSP [31,32].
2.5. Phylogenetic Analysis of STs
The internal fragments of the 7 housekeeping genes were concatenated using Snapgene (Version 4.2.4). Phylogenetic analysis of the concatenated sequences was performed by the neighbor-joining (NJ) method using MEGA X software with 1000 bootstrap replications to assess support for the various groups. The clonal complexes were identified by the goe BURST (Version 1.2.1) program according to the STs [33]. Split decomposition trees were generated by using splits tree (Version 4.18.1).
3. Results
3.1. MLST Analysis
The pmi, infB, mdh, adk, deoD, recA, and zwf genes were identified as the housekeeping genes of the Av. paragallinarum MLST scheme. All of these genes are present in a single copy in Av. paragallinarum genomes, and the length range of the fragments used for typing was 432–864 bp (Table 2). The average G+C content of the housekeeping gene fragments ranged from 44 to 48%. The number of polymorphic sites varied significantly among the seven housekeeping genes. The partial sequences of the genes adk and pmi revealed a minimum and maximum of 2 and 109 polymorphic sites, respectively. For 59 Av. paragallinarum strains, the number of alleles for the housekeeping genes was as follows: three (pmi and adk), four (mdh and deoD), five (recA and zwf), and six (infB). The nucleotide polymorphism is the average number of nucleotide differences between any two alleles in a population and is used to measure the degree of difference in genetic information in a population. The nucleotide polymorphisms of the seven housekeeping genes ranged from 0.00015 (adk) to 0.02823 (pmi). The dN/dS values ranged from 0 to 0.80, all less than 1. The Simpson’s index of diversity of the housekeeping genes ranged from 0.067 to 0.613, with adk having the lowest value of 0.067 and zwf having the highest value of 0.613. The neutral theory test results revealed that the D values of pmi, adk, and recA were all less than 0, while the D values of the remaining four genes were greater than 0. All the housekeeping genes did not deviate significantly from 0. These data are presented in Table 3.
Table 3.
Genetic diversity of housekeeping genes in 59 Av. paragallinarum strains.
The seven housekeeping genes of the strain 2019JS28 were taken as allele 1 for each gene, forming ST1. According to the nucleotide sequence analysis of the seven housekeeping genes, 59 strains of Av. paragallinarum were classified into eight ST types. The highest proportion was ST1 (34 strains) with 57.63% of the total number of isolates, followed by ST3 (11 strains) with 18.64% and ST8 (9 strains) with 15.25%. ST2, ST4, ST5, ST6, and ST7 each had only one strain (Table 4). Based on the above results, we applied to PubMLST for a public MLST site for Av. paragallinarum (https://pubmlst.org/organisms/avibacterium-paragallinarum, accessed on 21 March 2024).
Table 4.
STs of 59 Av. paragallinarum strains.
3.2. Phylogeny and Cluster Analysis of STs
After connecting the seven housekeeping genes in the order of pmi, infB, mdh, adk, deoD, recA, and zwf, the phylogenetic tree was constructed. As shown in Figure 1, three major branches were observed for the 59 Av. paragallinarum strains. Most of the isolates from China belonged to one group, with the exception of the 2021/06 and M strains, which showed a marked genetic distance from the other strains from China. All isolates from North America, including Mexico and the United States, belonged to the same branch. The genetic distance between the last branch and the above two evolutionary branches was relatively far, and it was further divided into two small evolutionary branches. The FARAPER-174 strain belonged to a small branch, and the other branch included the 2021/06, M, and H-18 strains. The Av. paragallinarum strains used for MLST analysis in this study were isolated between 2008 and 2021, as well as two reference strains 221 and H-18 from earlier years. Nineteen strains isolated in 2019, five strains in 2020, nine strains in 2021, and the reference strain 221 were assigned to ST1. Seven strains from 2019, one strain from 2015, and one strain from 2008 were assigned to ST8. In addition, ST1 contained serovars A and C, and ST3 was mainly serovar B. The serovars of ST8, ST4, and ST5 were unknown.
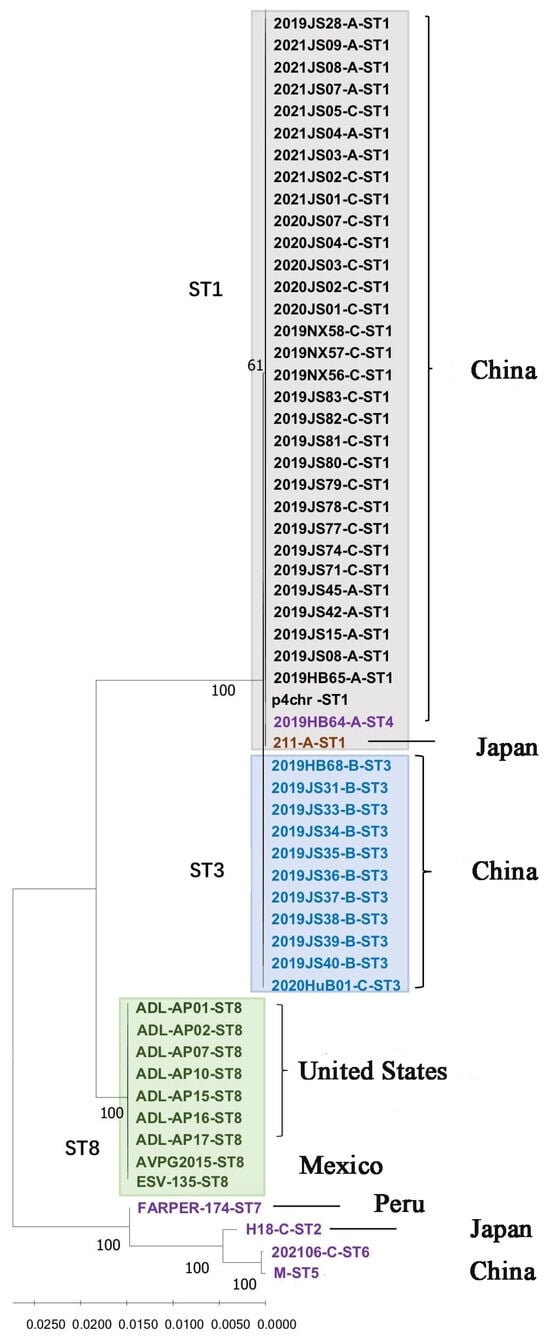
Figure 1.
Phylogenetic tree showing the relatedness of 59 Av. paragallinarum strains generated from MLST sequences.
The analytical evaluation of the clone group relationship between the STs showed that MLST formed two clone complexes (CC1 and CC2) and three singletons (ST2, ST5, and ST6) (Figure 2). CC1 was a dominant clone complex (47 strains) composed of three ST types: ST1, ST3, and ST4, with ST1 being the central ST type. CC2 was composed of ST7 and ST8. Among the 44 Av. paragallinarum strains isolated in China, 43 strains belonged to CC1, and two strains belonged to independent types ST5 and ST6, respectively. The strains of CC2 were all from abroad; ST8 from North America, and ST7 from Peru. This indicates that the genome sequences of the Av. paragallinarum isolated from China were consistent and had a distant genetic relationship with foreign CC2, indicating strong regional specificity.
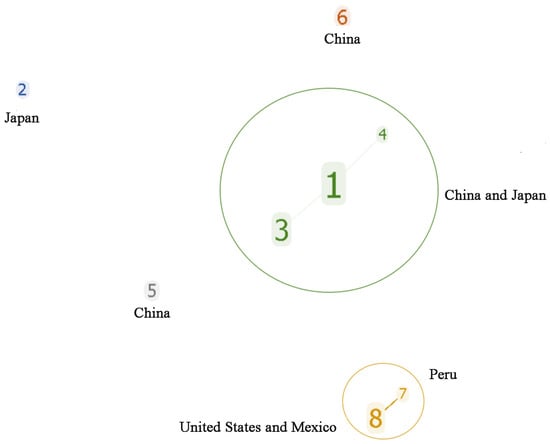
Figure 2.
Cluster analysis for STs of 59 Av. paragallinarum strains.
4. Discussion
The purpose of this study was to characterize Av. paragallinarum strains under standardized conditions by developing an MLST scheme. This scheme can be easily carried out by other laboratories and can compare Av. paragallinarum sequences globally. To achieve this idea, seven housekeeping genes were identified, and 59 Av. paragallinarum strains were sequenced and compared.
IC is a common infectious disease in chickens, characterized by acute onset and high infectivity. With the increasing intensification of chicken farms, the high bird density increases the pressure on the prevention and treatment of respiratory diseases such as IC [2]. At the same time, with the implementation of the drug reduction policy and the increase in the supervision and punishment of drug residues, antibiotics are strictly restricted, particularly during the laying period, which may be another reason for the rising incidence rate of IC in China [15,27]. Due to the lack of cross protection between different serovars, it is necessary to classify and identify epidemic strains to guide vaccine selection [34]. As mentioned earlier, the serological typing method based on HI test requires the standard serum of different serovars and can only be carried out by laboratories conducting relevant research [35]. Therefore, based on traditional bacterial isolation, there is an urgent need to develop a molecular typing method for Av. paragallinarum.
In this study, seven housekeeping genes (pmi, infB, mdh, adk, deoD, recA, and zwf) were used to establish the MLST method for typing Av. paragallinarum. These seven genes are relatively conserved and stable and are located throughout the chromosome of Av. paragallinarum in a relatively dispersed manner, which can avoid the influence of genetic linkage and reflect the phylogenetic relationship between different strains. To further demonstrate the feasibility of the housekeeping genes, the number of allele types, the number of polymorphic sites, nucleotide polymorphism, dN/dS, Simpson’s index, and neutral theory were evaluated. Zwf had the highest number of alleles, while adk and pmi had the lowest number of alleles. Simpson’s index is used to describe the probability that the number of individuals obtained from two consecutive samplings in a community belong to the same species. It is used to estimate the species diversity of the population. The Simpson’s index ranges from 0 to 1, with higher values indicating greater population diversity [36]. The Simpson’s index of zwf was the highest with 0.613, indicating the diversity of zwf. The number of polymorphic sites refers to differences in gene sequences at specific loci on the allele, which are considered to be early mutations that can be stably inherited and are indicative of the significance of the genetic evolution of the strain [37]. The polymorphism mutation rate reflects the degree of variation within the housekeeping genes, with the lowest mutation rate for adk ensuring the stability of MLST, and the highest for pmi ensuring the resolution of the MLST. The dN/dS values can determine the degree of selection pressure of the housekeeping genes [38]. The nucleotide polymorphism and dN/dS values indicate the stability of the housekeeping genes at the nucleotide and amino acid levels, respectively. In this study, the nucleotide polymorphism of the seven housekeeping genes was small (0.00015–0.02823), and the dN/dS values were all less than 1, indicating that the selected housekeeping genes met the requirements for conservation and stability. The neutral evolution theory suggests that biological evolution is mainly due to random genetic drift of neutral mutations in natural populations, independent of natural selection. The neutral theory test results showed that the values were close to 1, which proves that the selected housekeeping genes conformed to the neutral theory and were under little pressure from natural selection [32]. These findings indicate that the selected housekeeping genes can maintain their conservation and reflect the diversity of genetic site differences. The seven housekeeping genes had sufficient stability and resolution, which could meet the requirements for establishing the MLST method for Av. paragallinarum.
The MLST analysis included strains from three continents and five different countries that were widely distributed across STs and phylogenetic clusters. The MLST based on seven housekeeping genes divided 59 strains of Av. paragallinarum into eight ST types, which were clustered into three evolutionary branches. Most of the isolates from China belonged to ST1 and ST3 in CC1. However, the strains 2021/06 and M belonged to ST6 and ST5 respectively, which were genetically distant from CC1. There could be several reasons for this, including genetic mutation, introduction from overseas, or native diversity. These isolates may have accumulated enough mutations over time. Secondly, globalization allows pathogens to spread rapidly between countries, so these strains could be the result of international transmission. To determine this result, we analyzed additional epidemiological data. Tracing the origin of these two strains showed that strain M was submitted to GenBank without any isolation information. Similarly, the original source of strain 2021/06 was also unknown. Since the original source information of these two strains is uncertain, based on the MLST results of this study, we speculate that strains 2021/06 and M may not have been isolated in China. All isolates from North America, including Mexico and the United States, were ST8 and belonged to the same branch. ST7 from Peru and ST8 from North America together formed CC2. Av. paragallinarum strains belonging to the same ST type are generally from the same geographical origin, indicating that a specific ST is associated with a geographical origin. ST1 contained the serovars A and C, and ST3 was predominantly the serovar B, suggesting a possible correlation between serovars and phylogenetic relationships. Through analysis, MLST can understand the genetic and evolutionary characteristics of Av. paragallinarum strains, providing useful information for the prevention and treatment of IC. However, the conclusions drawn from the available data are still very limited, which is directly related to the limitations of our sample size. We uploaded this MLST scheme to create a public database for Av. paragallinarum, collecting sequences and isolates from around the world. With more samples and ST sequence data, the genetic evolution and epidemic patterns of Av. paragallinarum can be clearly observed.
5. Conclusions
The MLST scheme established in this study can provide a clear, reproducible, and portable typing system in order to gain insight into the population structure of Av. paragallinarum by identifying genetic variation in bacterial sequences. Eight STs were identified by MLST and clustered into three evolutionary branches. In general, Av. paragallinarum strains from the same geographical region were separated by MLST and assigned to identical STs. We hope that more people can enrich and improve this MLST database and use the typing technology as a technical reserve for Av. paragallinarum monitoring and evolutionary analysis. In the future, this method can monitor the epidemic trend of Av. paragallinarum through genetic evolutionary traceability and compare the correlation between epidemics at different times in the same area or at the same time in different areas, so as to achieve more accurate prevention and control.
Author Contributions
Conceptualization, M.G. and X.Z.; methodology, Y.J.; software, Y.J.; validation, H.W.; formal analysis, Y.J.; investigation, Y.J.; resources, Y.J.; data curation, H.W.; writing—original draft preparation, M.G.; writing—review and editing, M.G. and Y.W.; visualization, M.G.; supervision, Y.W.; project administration, X.Z.; funding acquisition, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Key R&D Program of China (2023YFD1800200); the China Agriculture Research System of MOF and MARA (CARS-40); a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Institutional Review Board Statement
All samples used in this study were collected from clinical samples provided by chicken farms. Ethics approval was not required.
Informed Consent Statement
Informed consent was obtained from the owners of the chicken farms involved in this study.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Blackall, P.J. Infectious coryza: Overview of the disease and new diagnostic options. Clin. Microbiol. Rev. 1999, 12, 627–632. [Google Scholar] [CrossRef]
- Blackall, P.J.; Soriano-Vargas, E. Infectious coryza and related bacterial infections. Dis. Poult. 2020, 890–906. [Google Scholar] [CrossRef]
- Blackall, P.J.; Morrow, C.J.; McInnes, A.; Eaves, L.E.; Rogers, D.G. Epidemiologic studies on infectious coryza outbreaks in northern New South Wales, Australia, using serotyping, biotyping, and chromosomal DNA restriction endonuclease analysis. Avian Dis. 1990, 34, 267–276. [Google Scholar] [CrossRef]
- Page, L.A. Haemophilus infections in chickens. I. Characteristics of 12 Haemophilus isolates recovered from diseased chickens. Am. J. Vet. Res. 1962, 23, 85–95. [Google Scholar] [PubMed]
- Kume, K.; Sawata, A.; Nakai, T.; Matsumoto, M. Serological classification of Haemophilus paragallinarum with a hemagglutinin system. J. Clin. Microbiol. 1983, 17, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Blackall, P.J.; Eaves, L.E.; Rogers, D.G. Proposal of a new serovar and altered nomenclature for Haemophilus paragallinarum in the Kume hemagglutinin scheme. J. Clin. Microbiol. 1990, 28, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Blackall, P.J.; Reid, G.G. Further efficacy studies on inactivated, aluminum-hydroxide-adsorbed vaccines against infectious coryza. Avian Dis. 1987, 31, 527–532. [Google Scholar] [CrossRef]
- Rimler, R.B.; Davis, R.B.; Page, R.K. Infectious coryza: Cross-protection studies, using seven strains of Haemophilus gallinarum. Am. J. Vet. Res. 1977, 38, 1587–1589. [Google Scholar] [PubMed]
- Kume, K.; Sawata, A.; Nakase, Y. Immunologic relationship between Page’s and Sawata’s serotype strains of Haemophilus paragallinarum. Am. J. Vet. Res. 1980, 41, 757–760. [Google Scholar]
- Yamaguchi, T.; Iritani, Y.; Hayashi, Y. Hemagglutinating activity and immunological properties of Haemophilus paragallinarum field isolates in Japan. Avian Dis. 1989, 33, 511–515. [Google Scholar] [CrossRef]
- Wang, H.; Sun, H.; Blackall, P.J.; Zhang, Z.; Zhou, H.; Xu, F.; Chen, X. Evaluation of a proposed molecular methodology for the serotyping of Avibacterium paragallinarum. J. Vet. Diagn. Invest. 2016, 28, 555–560. [Google Scholar] [CrossRef]
- Blackall, P.J.; Eaves, L.E.; Aus, G. Serotyping of Haemophilus paragallinarum by the Page scheme: Comparison of the use of agglutination and hemagglutination-inhibition tests. Avian Dis. 1990, 34, 643–645. [Google Scholar] [CrossRef]
- Zhang, P.J.; Miao, M.; Sun, H.; Gong, Y.; Blackall, P.J. Infectious coryza due to Haemophilus paragallinarum serovar B in China. Aust. Vet. J. 2003, 81, 96–97. [Google Scholar] [CrossRef]
- Sun, H.; Xie, S.; Li, X.; Xu, F.; Li, Y.; Boucher, C.E.; Chen, X. Selection of Avibacterium paragallinarum Page serovar B strains for an infectious coryza vaccine. Vet. Immunol. Immunopathol. 2018, 199, 77–80. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, J.; Huang, X.; Xu, M.; Feng, J.; Liu, C.; Zhang, G. Characterization of emergent Avibacterium paragallinarum strains and the protection conferred by infectious coryza vaccines against them in China. Poult. Sci. 2019, 98, 6463–6471. [Google Scholar] [CrossRef]
- Sakamoto, R.; Kino, Y.; Sakaguchi, M. Development of a multiplex PCR and PCR-RFLP method for serotyping of Avibacterium paragallinarum. J. Vet. Med. Sci. 2012, 74, 271–273. [Google Scholar] [CrossRef]
- Soriano, V.E.; Tellez, G.; Hargis, B.M.; Newberry, L.; Salgado-Miranda, C.; Vazquez, J.C. Typing of Haemophilus paragallinarum strains by using enterobacterial repetitive intergenic consensus-based polymerase chain reaction. Avian Dis. 2004, 48, 890–895. [Google Scholar] [CrossRef]
- Hellmuth, J.E.; Hitzeroth, A.C.; Bragg, R.R.; Boucher, C.E. Evaluation of the ERIC-PCR as a probable method to differentiate Avibacterium paragallinarum serovars. Avian Pathol. 2017, 46, 272–277. [Google Scholar] [CrossRef]
- Bisgaard, M.; Nørskov-Lauritsen, N.; de Wit, S.J.; Hess, C.; Christensen, H. Multilocus sequence phylogenetic analysis of Avibacterium. Microbiology 2012, 158 Pt 4, 993–1004. [Google Scholar] [CrossRef]
- Enright, M.C.; Spratt, B.G. Multilocus sequence typing. Trends Microbiol. 1999, 7, 482–487. [Google Scholar] [CrossRef]
- Selander, R.K.; Caugant, D.A.; Ochman, H.; Musser, J.M.; Gilmour, M.N.; Whittam, T.S. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl. Environ. Microbiol. 1986, 51, 873–884. [Google Scholar] [CrossRef]
- Grozner, D.; Kovacs, A.B.; Wehmann, E.; Kreizinger, Z.; Beko, K.; Mitter, A.; Sawicka, A.; Janosi, S.; Tomczyk, G.; Morrow, C.J.; et al. Multilocus sequence typing of the goose pathogen Mycoplasma anserisalpingitidis. Vet. Microbiol. 2021, 254, 108972. [Google Scholar] [CrossRef]
- Ibarz Pavon, A.B.; Maiden, M.C. Multilocus sequence typing. Methods Mol. Biol. 2009, 551, 129–140. [Google Scholar]
- Perez-Losada, M.; Cabezas, P.; Castro-Nallar, E.; Crandall, K.A. Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect. Genet. Evol. 2013, 16, 38–53. [Google Scholar] [CrossRef]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Guo, M.; Chen, X.; Zhang, H.; Liu, D.; Wu, Y.; Zhang, X. Isolation, Serovar Identification, and Antimicrobial Susceptibility of Avibacterium paragallinarum from Chickens in China from 2019 to 2020. Vet. Sci. 2022, 9, 27. [Google Scholar] [CrossRef]
- Chen, X.; Miflin, J.K.; Zhang, P.; Blackall, P.J. Development and Application of DNA Probes and PCR Tests for Haemophilus paragallinarum. Avian Dis. 1996, 40, 398–407. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carrico, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Soriano, E.V.; Garduño, M.L.; Téllez, G.; Rosas, P.F.; Suárez-Güemes, F.; Blackall, P.J. Cross-protection study of the nine serovars of Haemophilus paragallinarum in the Kume haemagglutinin scheme. Avian Pathol. 2004, 33, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B.; Zhang, L.; Koopman, J.S.; Manning, S.D.; Marrs, C.F. Choosing an appropriate bacterial typing technique for epidemiologic studies. Epidemiol. Perspect. Innov. 2005, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef] [PubMed]
- Coram, T.E.; Settles, M.L.; Wang, M.; Chen, X. Surveying expression level polymorphism and single-feature polymorphism in near-isogenic wheat lines differing for the Yr5 stripe rust resistance locus. Theor. Appl. Genet. 2008, 117, 401–411. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).