
Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases


Abstract
:Simple Summary
Abstract
1. Introduction
2. Classic Diagnostics versus Metagenomics
3. Advantages of Non-Targeted RNA-Based Metagenomics in Unveiling the Complex Microbial Landscape
4. Non-Targeted Nucleic Acid Sequencing as a Universal Diagnostic Approach across Animals, Plants, and the Environment
5. The Universality of Applications for Nucleic Acid-Based Non-Targeted Diagnostic Sequencing in Human Health
6. The Paradox of Non-Targeted NGS Applications for Clinical Diagnosis
7. The Need to Add Operational Value to NT-RNA-Seq
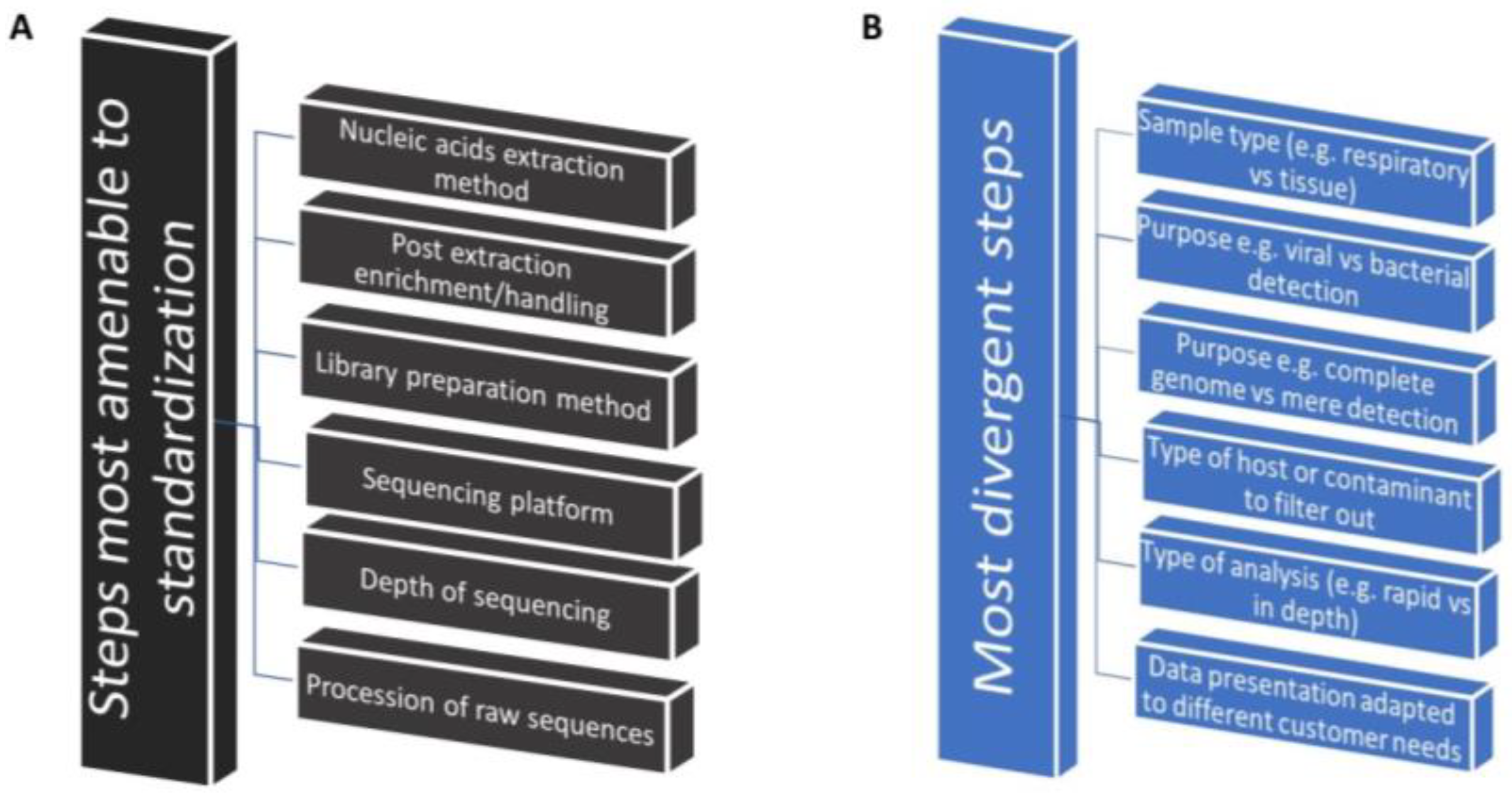
8. Need for Identification of the Best-Use Practices That Accelerate Implementation
8.1. Identification of Commonalities as the First Step toward Implementation Research
8.2. Comparative Studies on the Implementation
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Y.; Wang, G.; Lau, H.C.H.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, K.; Sekizuka, T.; Hashino, M.; Tanaka, R.; Kuroda, M. Disentangling Primer Interactions Improves SARS-CoV-2 Genome Sequencing by Multiplex Tiling PCR. PLoS ONE 2020, 15, e0239403. [Google Scholar] [CrossRef] [PubMed]
- Arana, C.; Liang, C.; Brock, M.; Zhang, B.; Zhou, J.; Chen, L.; Cantarel, B.; SoRelle, J.; Hooper, L.V.; Raj, P. A Short plus Long-Amplicon Based Sequencing Approach Improves Genomic Coverage and Variant Detection in the SARS-CoV-2 Genome. PLoS ONE 2022, 17, e0261014. [Google Scholar] [CrossRef] [PubMed]
- DNA Pipelines R&D; Farr, B.; Rajan, D.; Betteridge, E.; Shirley, L.; Quail, M.; Park, N.; Redshaw, N.; Bronner, I.; Aigrain, L.; et al. COVID-19 ARTIC v3 Illumina Library Construction and Sequencing Protocol. PLoS Glob. Public Health 2020. [Google Scholar] [CrossRef]
- Mitchell, S.L. Use of Diagnostic Metagenomics in the Clinical Microbiology Laboratory. Am. Soc. Clin. Lab. Sci. 2019, 35, 001768. [Google Scholar] [CrossRef]
- Pichler, I.; Schmutz, S.; Ziltener, G.; Zaheri, M.; Kufner, V.; Trkola, A.; Huber, M. Rapid and Sensitive Single-Sample Viral Metagenomics Using Nanopore Flongle Sequencing. J. Virol. Methods 2023, 320, 114784. [Google Scholar] [CrossRef] [PubMed]
- Pyatnitskiy, M.A.; Arzumanian, V.A.; Radko, S.P.; Ptitsyn, K.G.; Vakhrushev, I.V.; Poverennaya, E.V.; Ponomarenko, E.A. Oxford Nanopore Minion Direct Rna-Seq for Systems Biology. Biology 2021, 10, 1131. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Kim, D.; Kim, V.N.; Chang, H. Analyzing Viral Epitranscriptomes Using Nanopore Direct RNA Sequencing. J. Microbiol. 2022, 60, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Koonchanok, R.; Daulatabad, S.V.; Reda, K.; Janga, S.C. Sequoia: A Framework for Visual Analysis of RNA Modifications from Direct RNA Sequencing Data. Methods Mol Biol. 2023, 2624, 127–138. [Google Scholar] [CrossRef]
- Naarmann-De Vries, I.S.; Gjerga, E.; Gandor, C.L.A.; Dieterich, C. Adaptive Sampling for Nanopore Direct RNA-Sequencing. RNA 2023, 29, 1939–1949. [Google Scholar] [CrossRef]
- Ueda, H.; Dasgupta, B.; Yu, B.Y. RNA Modification Detection Using Nanopore Direct RNA Sequencing and NanoDoc2. Methods Mol. Biol. 2023, 2632, 299–319. [Google Scholar] [CrossRef] [PubMed]
- Al Kadi, M.; Okuzaki, D. Unfolding the Bacterial Transcriptome Landscape Using Oxford Nanopore Technology Direct RNA Sequencing. Methods Mol. Biol. 2023, 2632, 269–279. [Google Scholar] [CrossRef]
- Furlan, M.; Delgado-Tejedor, A.; Mulroney, L.; Pelizzola, M.; Novoa, E.M.; Leonardi, T. Computational Methods for RNA Modification Detection from Nanopore Direct RNA Sequencing Data. RNA Biol. 2021, 18, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Javaran, V.J.; Poursalavati, A.; Lemoyne, P.; Ste-Croix, D.T.; Moffett, P.; Fall, M.L. NanoViromics: Long-Read Sequencing of DsRNA for Plant Virus and Viroid Rapid Detection. Front. Microbiol. 2023, 14, 1192781. [Google Scholar] [CrossRef] [PubMed]
- Poursalavati, A.; Javaran, V.J.; Laforest-Lapointe, I.; Fall, M.L. Soil Metatranscriptomics: An Improved RNA Extraction Method Toward Functional Analysis Using Nanopore Direct RNA Sequencing. Phytobiomes J. 2023, 7, 42–54. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y.; Gao, G.F. Combining Metagenomics and Metatranscriptomics to Study Human, Animal and Environmental Resistomes. Med. Microecol. 2020, 3, 100014. [Google Scholar] [CrossRef]
- Zampieri, G.; Campanaro, S.; Angione, C.; Treu, L. Metatranscriptomics-Guided Genome-Scale Metabolic Modeling of Microbial Communities. Cell Rep. Methods 2023, 3, 100383. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinf. 2016, 12, 12s1. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Carda-Diéguez, M.; Aduse-Opoku, J.; Alsam, A.; Mira, A.; Curtis, M.A. The Murine Oral Metatranscriptome Reveals Microbial and Host Signatures of Periodontal Disease. J. Dent. Res. 2023, 102, 565–573. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Z.; Li, M.; Bartlam, M.; Wang, Y. Integrated Metagenomic and Metatranscriptomic Analysis Reveals Actively Expressed Antibiotic Resistomes in the Plastisphere. J. Hazard. Mater. 2022, 430, 128418. [Google Scholar] [CrossRef]
- Taj, B.; Adeolu, M.; Xiong, X.; Ang, J.; Nursimulu, N.; Parkinson, J. MetaPro: A Scalable and Reproducible Data Processing and Analysis Pipeline for Metatranscriptomic Investigation of Microbial Communities. Microbiome 2023, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, Y.; Liu, F.; Cao, J.; Lv, N.; Zhu, B.; Zhang, G.; Gao, G.F. Integrated Metagenomic and Metatranscriptomic Profiling Reveals Differentially Expressed Resistomes in Human, Chicken, and Pig Gut Microbiomes. Environ. Int. 2020, 138, 105649. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Knox, N.C.; Peterson, C.L.; Reimer, A.R. Highlighting Clinical Metagenomics for Enhanced Diagnostic Decision-Making: A Step Towards Wider Implementation. Comput. Struct. Biotechnol. J. 2018, 16, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Vonaesch, P.; Anderson, M.; Sansonetti, P.J. Pathogens, Microbiome and the Host: Emergence of the Ecological Koch’s Postulates. FEMS Microbiol. Rev. 2018, 42, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, G.; Cutler, S. Evolution of the Koch Postulates: Towards a 21st-Century Understanding of Microbial Infection. Clin. Microbiol. Infect. 2016, 22, 583–584. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, L.B.; Stanton, J.B.; Zhang, J.; Brown, C.; Butt, S.L.; Dimitrov, K.; Afonso, C.L.; Volkening, J.D.; Lara, L.J.C.; de Oliveira, C.S.F.; et al. Runting and Stunting Syndrome in Broiler Chickens: Histopathology and Association with a Novel Picornavirus. Vet. Pathol. 2021, 58, 123–135. [Google Scholar] [CrossRef]
- Wei, S.; Bahl, M.I.; Baunwall, S.M.D.; Hvas, C.L.; Licht, T.R. Determining Gut Microbial Dysbiosis: A Review of Applied Indexes for Assessment of Intestinal Microbiota Imbalances. Appl. Environ. Microbiol. 2021, 87, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pogranichniy, R.M.; Yoon, K.J.; Harms, P.A.; Sorden, S.D.; Daniels, M. Case-Control Study on the Association of Porcine Circovirus Type 2 and Other Swine Viral Pathogens with Postweaning Multisystemic Wasting Syndrome. J. Vet. Diagn. Investig. 2002, 14, 449–456. [Google Scholar] [CrossRef]
- Magro, D.O.; Santos, A.; Guadagnini, D.; de Godoy, F.M.; Silva, S.H.M.; Lemos, W.J.F.; Vitulo, N.; Torriani, S.; Pinheiro, L.V.; Martinez, C.A.R.; et al. Remission in Crohn’s Disease Is Accompanied by Alterations in the Gut Microbiota and Mucins Production. Sci. Rep. 2019, 9, 13263. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Prince, D.; Goh, K.G.K.; Katupitiya, L.; Gosling, D.; Crowley, M.R.; Crossman, D.K.; Ulett, G.C. Dual RNA Sequencing of Group B Streptococcus-Infected Human Monocytes Reveals New Insights into Host–Pathogen Interactions and Bacterial Evasion of Phagocytosis. Sci. Rep. 2023, 13, 2137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Z.; Zhang, J.; Zhang, Q.; Zheng, M.; Wen, J.; Zhao, G.; Li, Q. Dual RNA-Seq of H5N1 Avian Influenza Virus and Host Cell Transcriptomes Reveals Novel Insights Into Host-Pathogen Cross Talk. Front. Microbiol. 2022, 13, 828277. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Barquist, L.; Vogel, J. Resolving Host–Pathogen Interactions by Dual RNA-Seq. PLoS Pathog. 2017, 13, e1006033. [Google Scholar] [CrossRef] [PubMed]
- Seelbinder, B.; Wallstabe, J.; Marischen, L.; Weiss, E.; Wurster, S.; Page, L.; Löffler, C.; Bussemer, L.; Schmitt, A.L.; Wolf, T.; et al. Triple RNA-Seq Reveals Synergy in a Human Virus-Fungus Co-Infection Model. Cell Rep. 2020, 33, 108389. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Shinde, P.B.; Ahlawat, S.; Sharma, K.K. Understanding Virus–Bacteria–Human Tripartite Interactions: Strategies and Challenges. In Microbial Bioprocesses: Applications and Perspectives; Academic Press: Cambridge, MA, USA, 2023. [Google Scholar]
- Yadav, A.; Shamim, U.; Ravi, V.; Devi, P.; Kumari, P.; Maurya, R.; Das, P.; Somani, M.; Budhiraja, S.; Tarai, B.; et al. Early Transcriptomic Host Response Signatures in the Serum of Dengue Patients Provides Insights into Clinical Pathogenesis and Disease Severity. Sci. Rep. 2023, 13, 14170. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.; Hayward, R.; Shetty, A.; Mahurkar, A.; Humphrys, M.; Myers, G. Bioinformatic Analysis of Bacteria and Host Cell Dual RNA-Sequencing Experiments. Brief. Bioinform. 2017, 19, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.W.; Humphrys, M.S.; Myers, G.S.A. A Laboratory Methodology for Dual RNA-Sequencing of Bacteria and Their Host Cells in Vitro. Front. Microbiol. 2017, 8, 1830. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, S.; Visser, E.A.; Zwart, L.; du Toit, Y.; Bhadauria, V.; Shuey, L.S. Dual Rna-Seq to Elucidate the Plant–Pathogen Duel. Curr. Issues Mol. Biol. 2018, 27, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Di Gaspero, G.; Radovic, S.; De Luca, E.; Spadotto, A.; Magris, G.; Falginella, L.; Cattonaro, F.; Marroni, F. Evaluation of Sensitivity and Specificity in RNA-Seq-Based Detection of Grapevine Viral Pathogens. J. Virol. Methods 2022, 300, 114383. [Google Scholar] [CrossRef]
- Rey-Campos, M.; González-Vázquez, L.D.; Novoa, B.; Figueras, A. Metatranscriptomics Unmasks Mollusca Virome with a Remarkable Presence of Rhabdovirus in Cephalopods. Front. Mar. Sci. 2023, 10, 1209103. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, Y.; Qu, A.; Wang, J.; Zhao, J.; Ke, Q.; Chen, X.; Pu, F.; Wu, L.; Xu, P.; et al. Dual RNA-Seq Reveals a Host-Pathogen Interaction Transcriptional Regulation Pattern between Cryptocaryon irritans and Large Yellow Croaker (Larimichthys crocea). Aquacult. 2023, 565, 739104. [Google Scholar] [CrossRef]
- Afonso, C.L.; Afonso, A.M. Next-Generation Sequencing for the Detection of Microbial Agents in Avian Clinical Samples. Vet. Sci. 2023, 10, 690. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, S.; Huang, B.; Zhong, H.; Pan, Z.; Zhuang, Q.; Peng, C.; Hou, G.; Wang, K. Viral Infection Detection Using Metagenomics Technology in Six Poultry Farms of Eastern China. PLoS ONE 2019, 14, e0211553. [Google Scholar] [CrossRef]
- Kwoka, K.T.T.; de Rooij, M.M.T.; Messink, A.B.; Wouters, I.M.; Smit, L.A.M.; Heederik, D.J.J.; Koopmans, M.P.G.; Phan, M.V.T. Comparative Viral Metagenomics from Chicken Feces and Farm Dust in the Netherlands. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cibulski, S.; Alves de Lima, D.; Fernandes dos Santos, H.; Teixeira, T.F.; Tochetto, C.; Mayer, F.Q.; Roehe, P.M. A Plate of Viruses: Viral Metagenomics of Supermarket Chicken, Pork and Beef from Brazil. Virology 2021, 552, 1–9. [Google Scholar] [CrossRef]
- Boros, A.; Laszlo, Z.; Pankovics, P.; Marosi, A.; Albert, M.; Csagola, A.; Biro, H.; Fahsbender, E.; Delwart, E.; Reuter, G. High Prevalence, Genetic Diversity and a Potentially Novel Genotype of Sapelovirus A (Picornaviridae) in Enteric and Respiratory Samples in Hungarian Swine Farms. J. Gen. Virol. 2020, 101, 609–621. [Google Scholar] [CrossRef]
- Létourneau, V.; Gagné, M.-J.; Vyskocil, J.M.; Brochu, V.; Robitaille, K.; Gauthier, M.; Brassard, J.; Duchaine, C. Hunting for a Viral Proxy in Bioaerosols of Swine Buildings Using Molecular Detection and Metagenomics. J. Environ. Sci. 2023, 148, 69–78. [Google Scholar] [CrossRef]
- Ong, C.T.; Turni, C.; Blackall, P.J.; Boe-Hansen, G.; Hayes, B.J.; Tabor, A.E. Interrogating the Bovine Reproductive Tract Metagenomes Using Culture-Independent Approaches: A Systematic Review. Anim. Microbiome 2021, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A Metagenomics and Case-Control Study To Identify Viruses Associated with Bovine Respiratory Disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef]
- Ambrose, R.K.; Blakebrough-Hall, C.; Gravel, J.L.; Gonzalez, L.A.; Mahony, T.J. Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease. Viruses 2023, 15, 455. [Google Scholar] [CrossRef]
- Ahmadi, A.; Khezri, A.; Nørstebø, H.; Ahmad, R. A Culture-, Amplification-Independent, and Rapid Method for Identification of Pathogens and Antibiotic Resistance Profile in Bovine Mastitis Milk. Front. Microbiol. 2023, 13, 1104701. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Yan, Y. Metagenomic Analysis Reveals Antibiotic Resistance Genes in the Bovine Rumen. Microb. Pathog. 2020, 149, 104350. [Google Scholar] [CrossRef] [PubMed]
- Fried, W.A.; Soltero-Rivera, M.; Ramesh, A.; Lommer, M.J.; Arzi, B.; Derisi, J.L.; Horst, J.A. Use of Unbiased Metagenomic and Transcriptomic Analyses to Investigate the Association between Feline Calicivirus and Feline Chronic Gingivostomatitis in Domestic Cats. Am. J. Vet. Res. 2021, 82, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.S.; Wagner, J.; Kirkwood, C.D.; Gilkerson, J.R.; Mansfield, C.S. Characterization of the Fecal Virome in Dogs with Chronic Enteropathy. Vet. Microbiol. 2018, 221, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Liu, G.; Kalady, M.F.; Jin, T.; Ma, Y. Dysbiosis of the Stool DNA and RNA Virome in Crohn’s Disease. J. Med. Virol. 2023, 95, e28573. [Google Scholar] [CrossRef] [PubMed]
- Arroyo Mühr, L.S.; Dillner, J.; Ure, A.E.; Sundström, K.; Hultin, E. Comparison of DNA and RNA Sequencing of Total Nucleic Acids from Human Cervix for Metagenomics. Sci. Rep. 2021, 11, 18852. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.; Bassat, Q.; Medeiros, M.M.; Muñoz-Almagro, C. Respiratory Microbiota and Lower Respiratory Tract Disease. Expert Rev. Anti-Infect. Ther. 2017, 15, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Edgeworth, J.D. Respiratory Metagenomics: Route to Routine Service. Curr. Opin. Infect. Dis. 2023, 36, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Wylezich, C.; Höper, D. Meta-Ribosomalomics: RNA Sequencing Is an Unbiased Method for Parasite Detection of Different Sample Types. Front. Microbiol. 2021, 12, 614553. [Google Scholar] [CrossRef]
- Wylezich, C.; Calvelage, S.; Schlottau, K.; Ziegler, U.; Pohlmann, A.; Höper, D.; Beer, M. Next-Generation Diagnostics: Virus Capture Facilitates a Sensitive Viral Diagnosis for Epizootic and Zoonotic Pathogens Including SARS-CoV-2. Microbiome 2021, 9, 51. [Google Scholar] [CrossRef]
- Peterson, C.L.; Alexander, D.; Chen, J.C.Y.; Adam, H.; Walker, M.; Ali, J.; Forbes, J.; Taboada, E.; Barker, D.O.R.; Graham, M.; et al. Clinical Metagenomics Is Increasingly Accurate and Affordable to Detect Enteric Bacterial Pathogens in Stool. Microorganisms 2022, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Geng, S.; Mei, Q.; Zhang, L.; Yang, T.; Zhu, C.; Fan, X.; Wang, Y.; Tong, F.; Gao, Y.; et al. Diagnostic Value and Clinical Application of Metagenomic Next-Generation Sequencing for Infections in Critically Ill Patients. Infect. Drug Resist. 2023, 16, 6309–6322. [Google Scholar] [CrossRef]
- Saha, S.; Ramesh, A.; Kalantar, K.; Malaker, R.; Hasanuzzaman, M.; Khan, L.M.; Mayday, M.Y.; Sajib, M.S.I.; Li, L.M.; Langelier, C.; et al. Unbiased Metagenomic Sequencing for Pediatric Meningitis in Bangladesh Reveals Neuroinvasive Chikungunya Virus Outbreak and Other Unrealized Pathogens. mBio 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Sundararaju, S.; Tang, P.; Tsui, K.M.; Lopez, A.P.; Janahi, M.; Tan, R.; Tilley, P. A Metagenomics-Based Diagnostic Approach for Central Nervous System Infections in Hospital Acute Care Setting. Sci. Rep. 2020, 10, 11194. [Google Scholar] [CrossRef]
- Fan, G.; Li, S.; Tian, F.; Yang, L.; Yi, S.; Chen, S.; Li, C.; Zhang, R.; He, X.; Ma, X. RNA-Sequencing-Based Detection of Human Viral Pathogens in Cerebrospinal Fluid and Serum Samples from Children with Meningitis and Encephalitis. Microb. Genom. 2023, 9, 001079. [Google Scholar] [CrossRef]
- Shaw, A.G.; Cooper, L.V.; Gumede, N.; Bandyopadhyay, A.S.; Grassly, N.C.; Blake, I.M. Time Taken to Detect and Respond to Polio Outbreaks in Africa and the Potential Impact of Direct Molecular Detection and Nanopore Sequencing. J. Infect. Dis. 2022, 226, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.G.; Majumdar, M.; Troman, C.; O’Toole, Á.; Benny, B.; Abraham, D.; Praharaj, I.; Kang, G.; Sharif, S.; Alam, M.M.; et al. Rapid and Sensitive Direct Detection and Identification of Poliovirus from Stool and Environmental Surveillance Samples by Use of Nanopore Sequencing. J. Clin. Microbiol. 2020, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lizasoain, A.; Mir, D.; Masachessi, G.; Farías, A.; Rodríguez-Osorio, N.; Victoria, M.; Nates, S.; Colina, R. Environmental Surveillance through Next-Generation Sequencing to Unveil the Diversity of Human Enteroviruses beyond the Reported Clinical Cases. Viruses 2021, 13, 120. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Faye, M.; Diez-Fuertes, F.; Moreno-Docón, A.; Chirlaque-López, M.D.; Faye, O.; Cabrerizo, M. Metagenomic Sequencing, Molecular Characterization, and Bayesian Phylogenetics of Imported Type 2 Vaccine-Derived Poliovirus, Spain, 2021. Front. Cell. Infect. Microbiol. 2023, 13, 1168355. [Google Scholar] [CrossRef]
- Klapsa, D.; Wilton, T.; Zealand, A.; Bujaki, E.; Saxentoff, E.; Troman, C.; Shaw, A.G.; Tedcastle, A.; Majumdar, M.; Mate, R.; et al. Sustained Detection of Type 2 Poliovirus in London Sewage between February and July, 2022, by Enhanced Environmental Surveillance. Lancet 2022, 400, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.B.; Goyal, T.; Tyler, K.; Thakur, K.T. Re-Emergence of Poliovirus in the United States: Considerations and Implications. Ann. Neurol. 2022, 92, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Smibert, O.C.; Trubiano, J.A.; Slavin, M.A.; Kwong, J.C. An Infectious Diseases Perspective on the Microbiome and Allogeneic Stem Cell Transplant. Curr. Opin. Infect. Dis. 2020, 33, 426–432. [Google Scholar] [CrossRef]
- Ichiyama, T.; Nagai, Y.; Urushiyama, D.; Ohno, M.; Yamaguchi, T.; Nagayoshi, M.; Sakuraba, Y.; Yamasaki, F.; Kuroda, K.; Hata, K.; et al. New Relation between Dysbiosis of the Vaginal and Endometrial Microbiota and RIF Found. Fertil. Steril. 2019, 112, e333. [Google Scholar] [CrossRef]
- Wang, Q.; Miao, Q.; Pan, J.; Jin, W.; Ma, Y.; Zhang, Y.; Yao, Y.; Su, Y.; Huang, Y.; Li, B.; et al. The Clinical Value of Metagenomic Next-Generation Sequencing in the Microbiological Diagnosis of Skin and Soft Tissue Infections. Int. J. Infect. Dis. 2020, 100, 414–420. [Google Scholar] [CrossRef]
- Stockdale, S.R.; Blanchard, A.A.; Nayak, A.; Husain, A.; Nashine, R.; Dudani, H.; McClure, C.P.; Tarr, A.W.; Nag, A.; Meena, E.; et al. RNA-Seq of Untreated Wastewater to Assess COVID-19 and Emerging and Endemic Viruses for Public Health Surveillance. Lancet Reg. Health Southeast Asia 2023, 14, 100205. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Potential Applications of Human Viral Metagenomics and Reference Materials: Considerations for Current and Future Viruses. Appl. Environ. Microbiol. 2020, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Viral Metagenomics as a Tool to Track Sources of Fecal Contamination: A One Health Approach. Viruses 2023, 15, 236. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The Human Virome: Assembly, Composition and Host Interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef]
- Amrane, S.; Lagier, J.C. Metagenomic and Clinical Microbiology. Hum. Microb. J. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Batool, M.; Galloway-Peña, J. Clinical Metagenomics—Challenges and Future Prospects. Front. Microbiol. 2023, 14, 1186424. [Google Scholar] [CrossRef] [PubMed]
- Graff, K.; Dominguez, S.R.; Messacar, K. Metagenomic Next-Generation Sequencing for Diagnosis of Pediatric Meningitis and Encephalitis: A Review. J. Pediatric Infect. Dis. Soc. 2021, 10, S78–S87. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.H. We All Know Standardization Is Key, But How Do We Get There with Clinical Metagenomics? Clin. Chem. 2023, 69, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Gaston, D.C. Clinical Metagenomics for Infectious Diseases: Progress toward Operational Value. J. Clin. Microbiol. 2023, 61, e0126722. [Google Scholar] [CrossRef]
- Bihl, S.; de Goffau, M.; Podlesny, D.; Segata, N.; Shanahan, F.; Walter, J.; Fricke, W.F. When to Suspect Contamination Rather than Colonization–Lessons from a Putative Fetal Sheep Microbiome. Gut Microbes 2022, 14, 2005751. [Google Scholar] [CrossRef]
- Jurasz, H.; Pawłowski, T.; Perlejewski, K. Contamination Issue in Viral Metagenomics: Problems, Solutions, and Clinical Perspectives. Front. Microbiol. 2021, 12, 745076. [Google Scholar] [CrossRef] [PubMed]
- Zinter, M.S.; Mayday, M.Y.; Ryckman, K.K.; Jelliffe-Pawlowski, L.L.; Derisi, J.L. Towards Precision Quantification of Contamination in Metagenomic Sequencing Experiments. Microbiome 2019, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Bunholi, I.V.; Foster, N.R.; Casey, J.M. Environmental DNA and RNA in Aquatic Community Ecology: Toward Methodological Standardization. Environ. DNA 2023, 5, 1133–1147. [Google Scholar] [CrossRef]
- Kurian, S.M.; Gordon, S.; Barrick, B.; Dadlani, M.N.; Fanelli, B.; Cornell, J.B.; Head, S.R.; Marsh, C.L.; Case, J. Feasibility and Comparison Study of Fecal Sample Collection Methods in Healthy Volunteers and Solid Organ Transplant Recipients Using 16S RRNA and Metagenomics Approaches. Biopreserv. Biobank. 2020, 18, 425–440. [Google Scholar] [CrossRef]
- Maghini, D.G.; Dvorak, M.; Dahlen, A.; Roos, M.; Kuersten, S.; Bhatt, A.S. Quantifying Bias Introduced by Sample Collection in Relative and Absolute Microbiome Measurements. Nat. Biotechnol. 2023, 42, 328–338. [Google Scholar] [CrossRef]
- Zhang, X.L.; Deng, Y.P.; Yang, T.; Li, L.Y.; Cheng, T.Y.; Liu, G.H.; Duan, D.Y. Metagenomics of the Midgut Microbiome of Rhipicephalus Microplus from China. Parasit. Vectors 2022, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Rosseel, T.; Steensels, M.; van den Berg, T.; Lambrecht, B. What’s in a Strain? Viral Metagenomics Identifies Genetic Variation and Contaminating Circoviruses in Laboratory Isolates of Pigeon Paramyxovirus Type 1. Virus Res. 2013, 171, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Truchado, D.A.; Llanos-Garrido, A.; Oropesa-Olmedo, D.A.; Cerrada, B.; Cea, P.; Moens, M.A.J.; Gomez-Lucia, E.; Doménech, A.; Milá, B.; Pérez-Tris, J.; et al. Comparative Metagenomics of Palearctic and Neotropical Avian Cloacal Viromes Reveal Geographic Bias in Virus Discovery. Microorganisms 2020, 8, 1869. [Google Scholar] [CrossRef] [PubMed]
- Rosseel, T.; Ozhelvaci, O.; Freimanis, G.; Van Borm, S. Evaluation of Convenient Pretreatment Protocols for RNA Virus Metagenomics in Serum and Tissue Samples. J. Virol. Methods 2015, 222, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Schuele, L.; Lizarazo-Forero, E.; Strutzberg-Minder, K.; Schütze, S.; Löbert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.W.; Peter, S.; Rossen, J.W.A.; et al. Application of Shotgun Metagenomics Sequencing and Targeted Sequence Capture to Detect Circulating Porcine Viruses in the Dutch–German Border Region. Transbound. Emerg. Dis. 2022, 69, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Ogunbayo, A.E.; Sabiu, S.; Nyaga, M.M. Evaluation of Extraction and Enrichment Methods for Recovery of Respiratory RNA Viruses in a Metagenomics Approach. J. Virol. Methods 2023, 314, 114677. [Google Scholar] [CrossRef] [PubMed]
- Damian, D.; Maghembe, R.; Damas, M.; Wensman, J.J.; Berg, M. Application of Viral Metagenomics for Study of Emerging and Reemerging Tick-Borne Viruses. Vector-Borne Zoonotic Dis. 2020, 20, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Rosario-Cora, K. Enhancing Virus Surveillance through Metagenomics: Water Quality and Public Health Applications. Diss. Abstr. Int. Sect. B Sci. Eng. 2010. Available online: https://digitalcommons.usf.edu/etd/3600 (accessed on 9 May 2024).
- Fu, S.; Yang, Q.; Sheng, Y.; Wang, Q.; Wu, J.; Qiu, Z.; Lan, R.; Wang, Y.; Liu, Y. Metagenomics Combined with Comprehensive Validation as a Public Health Risk Assessment Tool for Urban and Agricultural Run-Off. Water Res. 2022, 209, 117941. [Google Scholar] [CrossRef]
- Ciulla, D.; Giannoukos, G.; Earl, A.; Feldgarden, M.; Gevers, D.; Levin, J.; Livny, J.; Ward, D.; Gnirke, A.; Nusbaum, C.; et al. Evaluation of Bacterial Ribosomal RNA (RRNA) Depletion Methods for Sequencing Microbial Community Transcriptomes. Genome Biol. 2010, 11, 9. [Google Scholar] [CrossRef]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by Poly (A) Capture, Ribosomal RNA Depletion, and DNA Microarray for Expression Profiling. Vector-Borne Zoonotic Dis. 2014, 15, 419. [Google Scholar] [CrossRef] [PubMed]
- Culviner, P.H.; Guegler, C.K.; Laub, M.T. A Simple, Cost-Effective, and Robust Method for rRNA Depletion in RNA-Sequencing Studies. mBio 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, A.; Oliveri, C.; Lucchi, A.; Pasquali, F.; Manfreda, G. Application of Shotgun Metagenomics to Smoked Salmon Experimentally Spiked: Comparison between Sequencing and Microbiological Data Using Different Bioinformatic Approaches. Ital. J. Food Saf. 2019, 8, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Fu, Q.; Winand, R.; Vanneste, K.; Hakhverdyan, M.; Höper, D.; Vandenbussche, F. Evaluation of a Commercial Exogenous Internal Process Control for Diagnostic RNA Virus Metagenomics from Different Animal Clinical Samples. J. Virol. Methods 2020, 283, 113916. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Steensels, M.; Mathijs, E.; Vandenbussche, F.; van den Berg, T.; Lambrecht, B. Metagenomic Sequencing Determines Complete Infectious Bronchitis Virus (Avian Gammacoronavirus) Vaccine Strain Genomes and Associated Viromes in Chicken Clinical Samples. Virus Genes 2021, 57, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Roupie, V.; Linden, A.; Vangeluwe, D.; De Waele, V.; Lambrecht, B.; Steensels, M. RNA Sequencing of Avian Paramyxovirus (Paramyxoviridae, Avulavirinae) Isolates from Wild Mallards in Belgium, 2021: Complete Genomes and Coinfections. Virus Genes 2023, 59, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, D.; Ji, Z.; Zhang, Y.; Wang, Y.; Chen, X.; He, Y.; Lu, X.; Li, R.; Guo, Y.; et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses 2022, 14, 2048. [Google Scholar] [CrossRef] [PubMed]
- Ibañez-Lligoña, M.; Colomer-Castell, S.; González-Sánchez, A.; Gregori, J.; Campos, C.; Garcia-Cehic, D.; Andrés, C.; Piñana, M.; Pumarola, T.; Rodríguez-Frias, F.; et al. Bioinformatic Tools for NGS-Based Metagenomics to Improve the Clinical Diagnosis of Emerging, Re-Emerging and New Viruses. Viruses 2023, 15, 587. [Google Scholar] [CrossRef] [PubMed]
- Gihawi, A.; Cardenas, R.; Hurst, R.; Brewer, D.S. Quality Control in Metagenomics Data. In Metagenomic Data Analysis; Springer: New York, NY, USA, 2023; Volume 2649. [Google Scholar] [CrossRef]
- Gemler, B.T.; Mukherjee, C.; Howland, C.; Fullerton, P.A.; Spurbeck, R.R.; Catlin, L.A.; Smith, A.; Minard-Smith, A.T.; Bartling, C. UltraSEQ, a Universal Bioinformatic Platform for Information-Based Clinical Metagenomics and Beyond. Microbiol. Spectr. 2023, 11, e0416022. [Google Scholar] [CrossRef]
- Grundy, B.S.; Parikh, H.; Jacob, S.; Banura, P.; Moore, C.C.; Liu, J.; Houpt, E.R. Pathogen Detection Using Metagenomic Next-Generation Sequencing of Plasma Samples from Patients with Sepsis in Uganda. Microbiol. Spectr. 2023, 11, e0431222. [Google Scholar] [CrossRef]
- Sichtig, H.; Minogue, T.; Yan, Y.; Stefan, C.; Hall, A.; Tallon, L.; Sadzewicz, L.; Nadendla, S.; Klimke, W.; Hatcher, E.; et al. FDA-ARGOS Is a Database with Public Quality-Controlled Reference Genomes for Diagnostic Use and Regulatory Science. Nat. Commun. 2019, 10, 3313. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Qi, C.; Yang, H.; Lu, M.; Cai, Y.; Fu, T.; Ren, J.; Jin, Q.; Zhang, X. GutMGene: A Comprehensive Database for Target Genes of Gut Microbes and Microbial Metabolites. Nucleic Acids Res. 2022, 50, D795–D800. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Sivabalan, S.K.M.; Mahesh, A.; Palanikumar, I.; Kuppa Baskaran, D.K.; Raman, K. Big Data for a Small World: A Review on Databases and Resources for Studying Microbiomes. J. Indian Inst. Sci. 2023, 103, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.K.; Hysom, D.A.; Gardner, S.N.; Lloyd, G.S.; Gokhale, M.B.; Allen, J.E. Scalable Metagenomic Taxonomy Classification Using a Reference Genome Database. Bioinformatics 2013, 29, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Rendón, J.M.; Lang, B.; Llorens, M.R.; Tartaglia, G.G.; Burgas, M.T. DualSeqDB: The Host-Pathogen Dual RNA Sequencing Database for Infection Processes. Nucleic Acids Res. 2021, 49, D687–D693. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.L.A.; Li, F.; Ghoshal, B.; McAllister, T.; Guan, L.L. Enhancing the Resolution of Rumen Microbial Classification from Metatranscriptomic Data Using Kraken and Mothur. Front. Microbiol. 2017, 8, 2445. [Google Scholar] [CrossRef] [PubMed]
- Sichtig, H.; Minogue, T.; Yan, Y.; Stefan, C.; Hall, A.; Tallon, L.; Sadzewicz, L.; Nadendla, S.; Klimke, W.; Hatcher, E.; et al. FDA-ARGOS: A Public Quality-Controlled Genome Database Resource for Infectious Disease Sequencing Diagnostics and Regulatory Science Research. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. Ultrafast and Accurate 16S RRNA Microbial Community Analysis Using Kraken 2. Microbiome 2020, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Terrón-Camero, L.C.; Gordillo-González, F.; Salas-Espejo, E.; Andrés-León, E. Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice. Genes 2022, 13, 2280. [Google Scholar] [CrossRef]
- Liebhoff, A.M.; Menden, K.; Laschtowitz, A.; Franke, A.; Schramm, C.; Bonn, S. Pathogen Detection in RNA-Seq Data with Pathonoia. BMC Bioinf. 2023, 24, 53. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, C.J.; Balan, P.; Suriyanarayanan, T.; Lakshmanan, M.; Lee, D.Y.; Rho, M.; Jakubovics, N.; Brandt, B.; Crielaard, W.; Zaura, E. Oral Microbiome-Systemic Link Studies: Perspectives on Current Limitations and Future Artificial Intelligence-Based Approaches. Crit. Rev. Microbiol. 2020, 46, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Yu, X.; Chen, Z. Applying Artificial Intelligence in the Microbiome for Gastrointestinal Diseases: A Review. J. Gastroenterol. Hepatol. 2021, 36, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Levitte, S.; Patel, A.; Balabanis, T.; Wei, M.T.; Sinha, S.R. Artificial Intelligence Applications in Inflammatory Bowel Disease: Emerging Technologies and Future Directions. World J. Gastroenterol. 2021, 27, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.K.; Roy, P.; Kumar, V.; ul Gani Mir, T. Metagenomics and Artificial Intelligence in the Context of Human Health. Infect. Genet. Evol. 2022, 100, 105267. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Mallet, L.; Blümel, J.; Cassart, J.P.; Knezevic, I.; Ng, S.H.S.; Wall, M.; Jakava-Viljanen, M.; Logvinoff, C.; Goios, A.; et al. Report of the Third Conference on Next-Generation Sequencing for Adventitious Virus Detection in Biologics for Humans and Animals. Biologicals 2023, 83, 101696. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Blümel, J.; Deforce, D.; Gruber, M.F.; Jungbäck, C.; Knezevic, I.; Mallet, L.; Mackay, D.; Matthijnssens, J.; O’Leary, M.; et al. Report of the Second International Conference on Next Generation Sequencing for Adventitious Virus Detection in Biologics for Humans and Animals. Biologicals 2020, 67, 94–111. [Google Scholar] [CrossRef]
- Mason, C.E.; Afshinnekoo, E.; Tighe, S.; Wu, S.; Levy, S. International Standards for Genomes, Transcriptomes, and Metagenomes. J. Biomol. Tech. JBT 2017, 28, 8–18. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| 1. Always use “standard operating procedures” for sample-to-sample comparison purposes. | |
| 2. Develop “a priori” a sampling strategy focused on the specific problem with the help of a field veterinarian and pathologist. | |
| 3. Develop a sampling strategy that covers “completely and evenly” the areas or the host of interest. | |
| 4. Minimize contamination from operators, non-target tissues, and the environment at all stages of collection. | |
| 5. Minimize post-sampling contamination; use masks, sterile plasticware, media, and antibiotics, if possible, for manipulation and storage. | |
| 6. Do not mix different types of samples (e.g., cloacal samples will dilute respiratory samples with bacterial nucleic acids). | |
| 7. Obtain sufficient starting sample material (RNA/DNA) to minimize the amplification steps (e.g., pool the same type of samples if necessary). | |
| 8. Minimize degradation of nucleic acids (RNAs are very sensitive) by using gloves, cold chains, and RNAse-free reagents. | |
| 9. Use trained operators at all stages of the process. | |
| 10. Use fast and reliable labeling (printed tags, barcoding, spreadsheets, instead of pens at the site). | |
| 11. Obtain and link the most complete metadata possible in all samples (e.g., farm clinical and management information). | |
| 12. Note “all’ clinical details associated with the host pathology for each sample. | |
| 13. When spotting on FTA cards, rigorously follow the recommendations on expiration dates, spotting volumes, drying time storage, and shipment conditions. | |
| 14. Include information in “the shipping form” that will be used for the interpretation of complex results such as: | |
| Date of collection, the name of the operator, and/or sample contact information. | |
| Flock identification (can be coded for confidentiality). | |
| Type of sample (oropharyngeal, cloacal, tissue). | |
| Species and age of the sampled birds. | |
| Optional information: vaccination; suspected disease; clinical lesions; histology; flock health; production problems; GPS location. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spatz, S.; Afonso, C.L. Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases. Vet. Sci. 2024, 11, 239. https://doi.org/10.3390/vetsci11060239
Spatz S, Afonso CL. Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases. Veterinary Sciences. 2024; 11(6):239. https://doi.org/10.3390/vetsci11060239
Chicago/Turabian StyleSpatz, Stephen, and Claudio L. Afonso. 2024. "Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases" Veterinary Sciences 11, no. 6: 239. https://doi.org/10.3390/vetsci11060239
APA StyleSpatz, S., & Afonso, C. L. (2024). Non-Targeted RNA Sequencing: Towards the Development of Universal Clinical Diagnosis Methods for Human and Veterinary Infectious Diseases. Veterinary Sciences, 11(6), 239. https://doi.org/10.3390/vetsci11060239