
Manipulation of Innate Immunity for Cancer Therapy in Dogs

Abstract
:1. Introduction
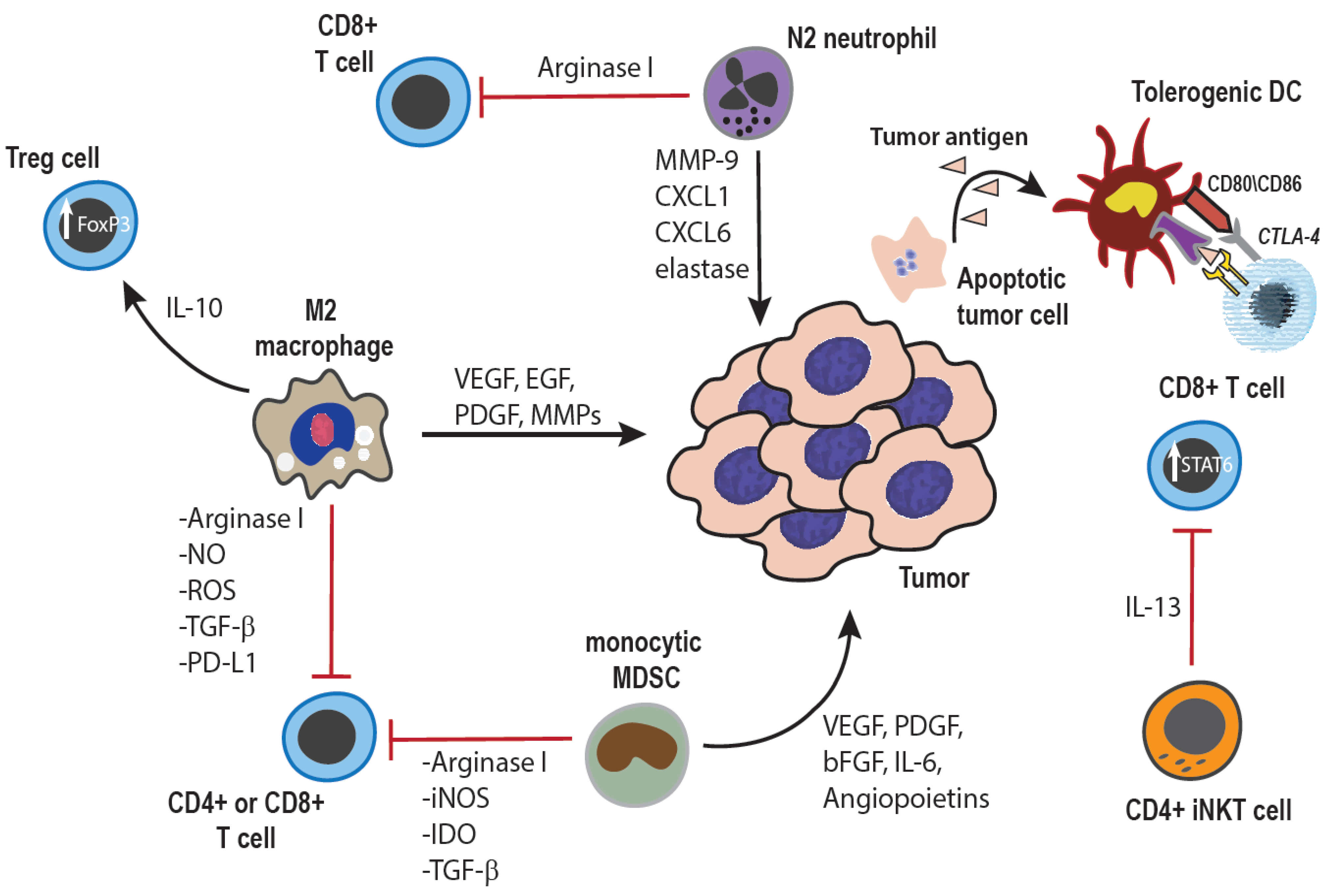
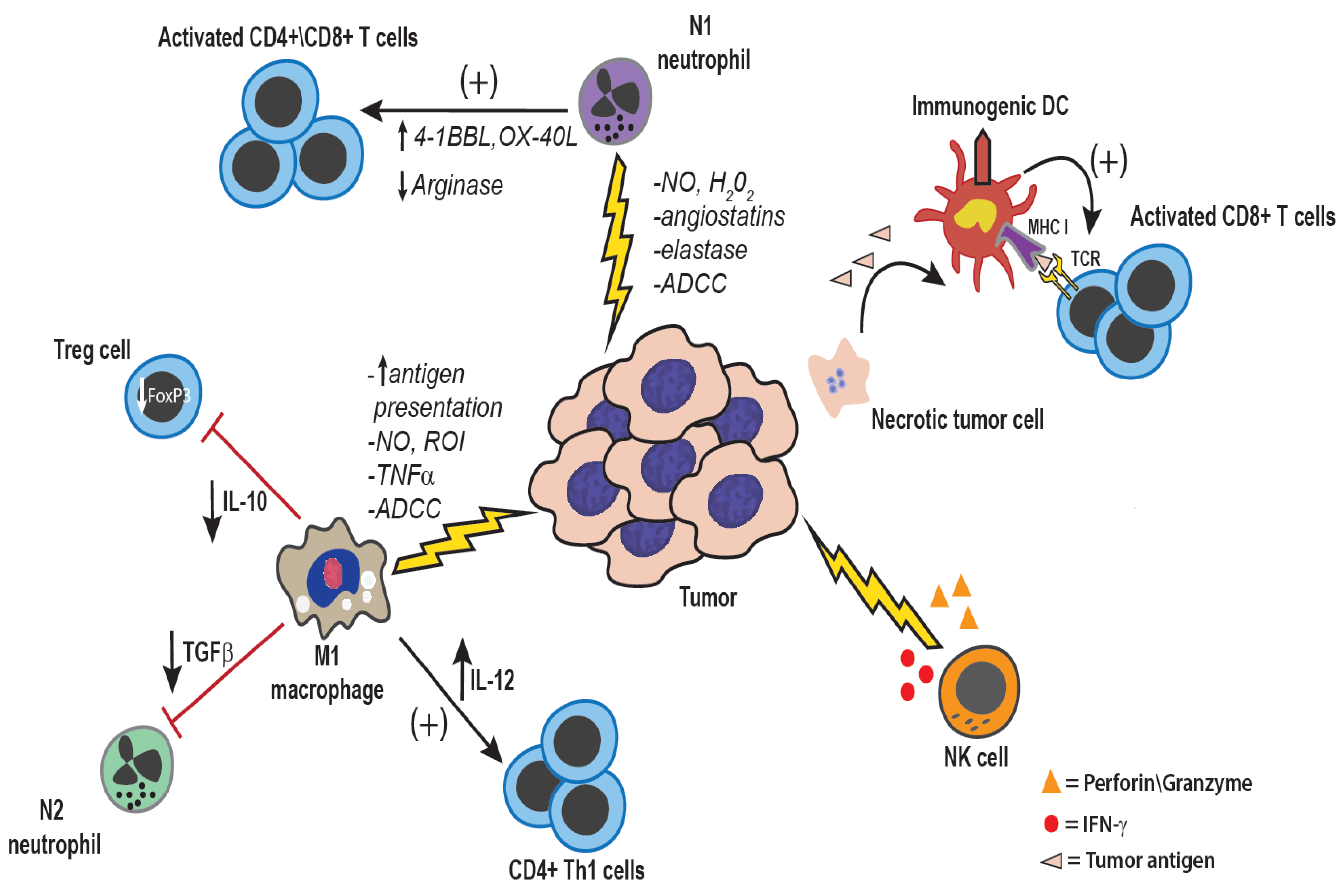
2. Dual Roles of Innate Immunity in Cancer Control



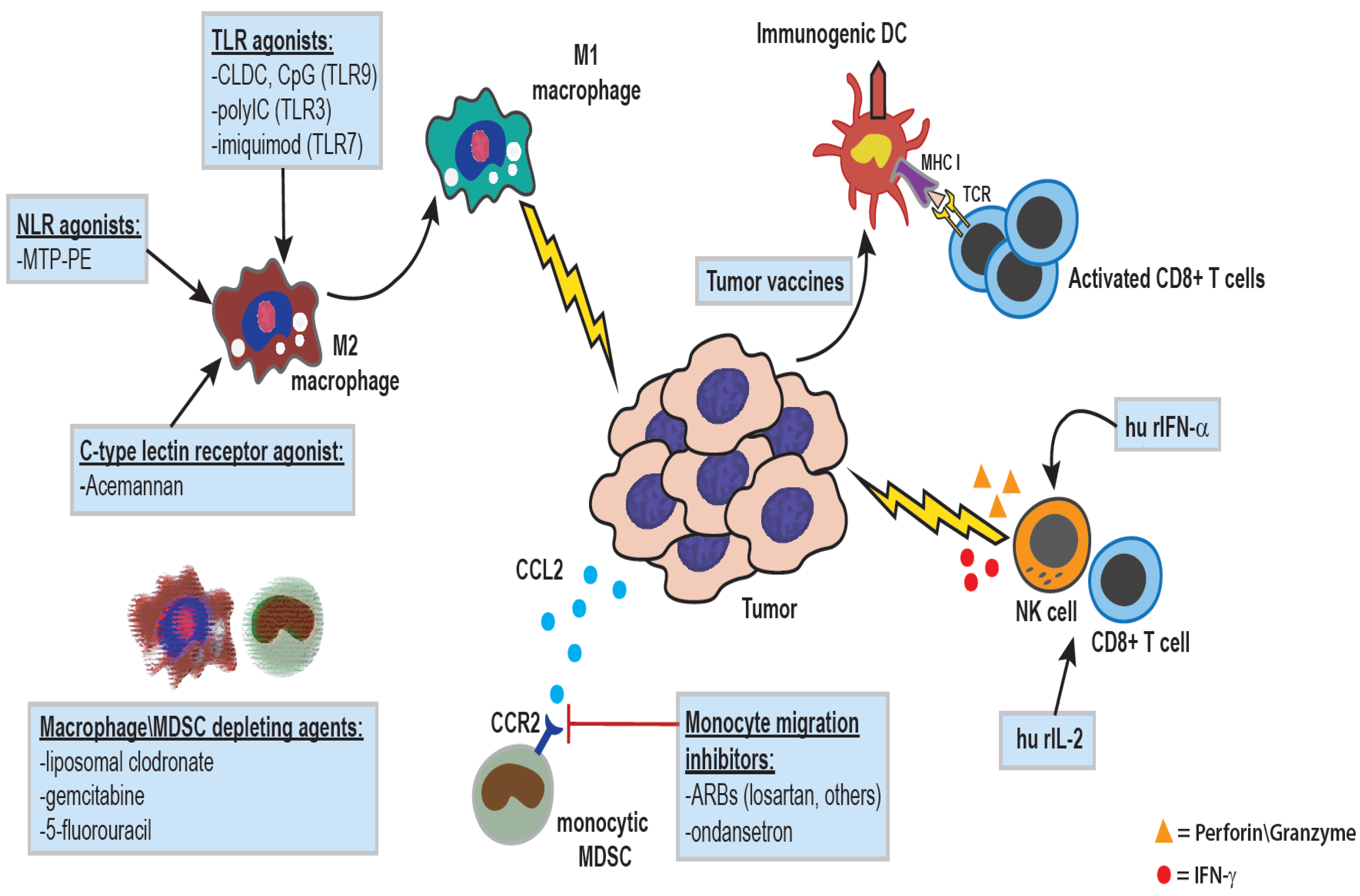
3. Activation of Innate Immunity for Cancer Immunotherapy Using Biological Response Modifiers
4. Activation of Innate Immunity by Nod-Like Receptor Antagonists
5. Activation of Anti-Tumor Immunity by Biological Molecules
6. Immunotherapy by Activation of C-Type Lectin Receptors Using Plant Extracts
7. Activation of Anti-Tumor Immunity in Dogs with TLR Agonists

{kind=link}
{kind=link}
{kind=link}
| Cytokine\Immune Molecule | Cellular Source | Anti-Tumor Mechanism(s) |
|---|---|---|
| IFN-γ |
|
|
| IL-12 |
| |
| IL-2 |
| |
| IFN-α/β |
|
|
| TNF-α |
|
|
| Perforin and granzyme |
|
|
| FasFasL |
| |
| TRAIL |
|
|
8. Innate Immune Activation by Recombinant Cytokines
9. Reversal of Immune Suppression by Macrophage Depletion or Monocyte Migration Blockade
10. Conclusions and Implications
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pardoll, D. Cancer and the immune system: Basic concepts and targets for intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Rao, S.; Lafferty, M.H.; Thamm, D.H.; Morley, P.S.; Withrow, S.J.; Dow, S.W. Association of blood monocyte and lymphocyte count and disease-free interval in dogs with osteosarcoma. J. Vet. Intern. Med. 2010, 24, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Thamm, D.H.; Eickhoff, J.; Avery, A.C.; Dow, S.W. Increased monocyte chemotactic protein-1 concentration and monocyte count independently associate with a poor prognosis in dogs with lymphoma. Vet. Comp. Oncol. 2011, 9, 55–64. [Google Scholar] [CrossRef] [PubMed]
- MacEwen, E.G.; Kurzman, I.D.; Rosenthal, R.C.; Smith, B.W.; Manley, P.A.; Roush, J.K.; Howard, P.E. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J. Natl. Cancer Inst. 1989, 81, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Kurzman, I.D.; MacEwen, E.G.; Rosenthal, R.C.; Fox, L.E.; Keller, E.T.; Helfand, S.C.; Vail, D.M.; Dubielzig, R.R.; Madewell, B.R.; Rodriguez, C.O., Jr. Adjuvant therapy for osteosarcoma in dogs: Results of randomized clinical trials using combined liposome-encapsulated muramyl tripeptide and cisplatin. Clin. Cancer Res. 1995, 1, 1595–1601. [Google Scholar] [PubMed]
- Janotova, T.; Jalovecka, M.; Auerova, M.; Švecová, I.; Bruzlová, P.; Maierová, V.; Kumžáková, Z.; Cunátová, Š.; Vlčková, Z.; Caisová, V.; et al. The use of anchored agonists of phagocytic receptors for cancer immunotherapy: B16-F10 murine melanoma model. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol. Immunother. 2002, 51, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 2006, 55, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Sherger, M.; Kisseberth, W.; London, C.; Olivo-Marston, S.; Papenfuss, T.L. Identification of myeloid derived suppressor cells in the peripheral blood of tumor bearing dogs. BMC Vet. Res. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Goulart, M.R.; Pluhar, G.E.; Ohlfest, J.R. Identification of myeloid derived suppressor cells in dogs with naturally occurring cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Maenhout, S.K.; Thielemans, K.; Aerts, J.L. Location, location, location: Functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Gagliani, N.; Huber, S.; Flavell, R.A. Innate immune cells in inflammation and cancer. Cancer Immunol. Res. 2013, 1, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” vs. “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Massari, F.; Amantini, C.; Nabissi, M.; Maines, F.; Burattini, L.; Berardi, R.; Santoni, G.; Montironi, R.; Tortora, G.; et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2015. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Larmonier, N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. 2014, 74, 2663–2668. [Google Scholar] [CrossRef] [PubMed]
- Tarek, N.; Lee, D.A. Natural killer cells for osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 341–353. [Google Scholar] [PubMed]
- Mentlik James, A.; Cohen, A.D.; Campbell, K.S. Combination immune therapies to enhance anti-tumor responses by NK cells. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Dunn, G.P.; Lacoursiere, D.Y.; Schreiber, R.D.; Bui, J.D. Cancer immunoediting of the NK group 2D ligand H60a. J. Immunol. 2011, 187, 3538–3545. [Google Scholar] [CrossRef] [PubMed]
- Berzofsky, J.A.; Terabe, M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunol. Immunother. 2008, 57, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Terabe, M.; Donaldson, D.D.; Forni, G.; Berzofsky, J.A. Natural immunosurveillance against spontaneous, autochthonous breast cancers revealed and enhanced by blockade of IL-13-mediated negative regulation. Cancer Immunol. Immunother. 2008, 57, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Hegmans, J.P.; Aerts, J.G. Immunomodulation in cancer. Curr. Opin. Pharmacol. 2014, 17, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hawes, M.C.; Wen, F.; Elquza, E. Extracellular DNA: A bridge to cancer. Cancer Res. 2015, 75, 4260–4264. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Dr William Coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [PubMed]
- Owen, L.N.; Bostock, D.E. Effects of intravenous BCG in normal dogs and in dogs with spontaneous osteosarcoma. Eur. J. Cancer 1974, 10, 775–780. [Google Scholar] [CrossRef]
- Owen, L.N.; Bostock, D.E.; Lavelle, R.B. Studies on chemotherapy and immunotherapy in canine lymphosarcoma and osteosarcoma. Bibl. Haematol. 1975, 43, 522–523. [Google Scholar] [PubMed]
- Bacon, N.J.; Ehrhart, N.P.; Dernell, W.S.; Lafferty, M.; Withrow, S.J. Use of alternating administration of carboplatin and doxorubicin in dogs with microscopic metastases after amputation for appendicular osteosarcoma: 50 cases (1999–2006). J. Am. Vet. Med. Assoc. 2008, 232, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Weinstein, M.J.; Springfield, D.S.; Rand, W.M. Results of surgery and doxorubicin chemotherapy in dogs with osteosarcoma. J. Am. Vet. Med. Assoc. 1995, 206, 1555–1560. [Google Scholar] [PubMed]
- Saam, D.E.; Liptak, J.M.; Stalker, M.J.; Chun, R. Predictors of outcome in dogs treated with adjuvant carboplatin for appendicular osteosarcoma: 65 cases (1996–2006). J. Am. Vet. Med. Assoc. 2011, 238, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, E.S.; Maeda, M.; Jaffe, N. Liposome-encapsulated muramyl tripeptide: A new biologic response modifier for the treatment of osteosarcoma. Cancer Treat Res. 1993, 62, 101–107. [Google Scholar] [PubMed]
- Kleinerman, E.S.; Jia, S.F.; Griffin, J.; Seibel, N.L.; Benjamin, R.S.; Jaffe, N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: The cytokine cascade and monocyte activation following administration. J. Clin. Oncol. 1992, 10, 1310–1316. [Google Scholar] [PubMed]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.R.; Beck, P.L.; Muruve, D.A. Nucleotide-binding oligomerization domain-like receptors and inflammasomes in the pathogenesis of non-microbial inflammation and diseases. J. Innate Immun. 2012, 4, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Kurzman, I.D.; Shi, F.; MacEwen, E.G. In vitro and in vivo canine mononuclear cell production of tumor necrosis factor induced by muramyl peptides and lipopolysaccharide. Vet. Immunol. Immunopathol. 1993, 38, 45–56. [Google Scholar] [CrossRef]
- MacEwen, E.G.; Kurzman, I.D.; Vail, D.M.; Dubielzig, R.R.; Everlith, K.; Madewell, B.R.; Rodriguez, C.O., Jr.; Phillips, B.; Zwahlen, C.H.; Obradovich, J.; et al. Adjuvant therapy for melanoma in dogs: Results of randomized clinical trials using surgery, liposome-encapsulated muramyl tripeptide, and granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 1999, 5, 4249–4258. [Google Scholar] [PubMed]
- Ishii, K.J.; Coban, C.; Akira, S. Manifold mechanisms of Toll-like receptor-ligand recognition. J. Clin. Immunol. 2005, 25, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Pierce, K.; King, G.; Yates, K.M.; Hall, J.; Tizard, I. Efficacy of acemannan in treatment of canine and feline spontaneous neoplasms. Mol. Biother. 1991, 3, 207–213. [Google Scholar] [PubMed]
- King, G.K.; Yates, K.M.; Greenlee, P.G.; Pierce, K.R.; Ford, C.R.; McAnalley, B.H.; Tizard, I.R. The effect of Acemannan Immunostimulant in combination with surgery and radiation therapy on spontaneous canine and feline fibrosarcomas. J. Am. Anim. Hosp. Assoc. 1995, 31, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. CpG DNA in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 2000, 247, 157–170. [Google Scholar] [PubMed]
- Walker, P.S.; Scharton-Kersten, T.; Krieg, A.M.; Love-Homan, L.; Rowton, E.D.; Udey, M.C.; Vogel, J.C. Immunostimulatory oligodeoxynucleotides promote protective immunity and provide systemic therapy for leishmaniasis via IL-12- and IFN-gamma-dependent mechanisms. Proc. Natl. Acad. Sci. USA 1999, 96, 6970–6975. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. DNA-based immune enhancers. Curr. Opin. Drug Discov. Devel. 2000, 3, 214–221. [Google Scholar] [PubMed]
- Jahrsdorfer, B.; Weiner, G.J. CpG oligodeoxynucleotides for immune stimulation in cancer immunotherapy. Curr. Opin. Investig. Drugs 2003, 4, 686–690. [Google Scholar] [PubMed]
- Marabelle, A.; Kohrt, H.; Caux, C.; Levy, R. Intratumoral immunization: A new paradigm for cancer therapy. Clin. Cancer Res. 2014, 20, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.W.; Fradkin, L.G.; Liggitt, D.H.; Willson, A.P.; Heath, T.D.; Potter, T.A. Lipid-DNA complexes induce potent activation of innate immune responses and antitumor activity when administered intravenously. J. Immunol. 1999, 163, 1552–1561. [Google Scholar] [PubMed]
- Dow, S. Liposome-nucleic acid immunotherapeutics. Expert Opin. Drug Deliv. 2008, 5, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.; Elmslie, R.; Kurzman, I.; MacEwen, G.; Pericle, F.; Liggitt, D. Phase I study of liposome-DNA complexes encoding the interleukin-2 gene in dogs with osteosarcoma lung metastases. Hum. Gene Ther. 2005, 16, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Kamstock, D.; Guth, A.; Elmslie, R.; Kurzman, I.; Liggitt, D.; Coro, L.; Fairman, J.; Dow, S. Liposome-DNA complexes infused intravenously inhibit tumor angiogenesis and elicit antitumor activity in dogs with soft tissue sarcoma. Cancer Gene Ther. 2006, 13, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, G.K.; Straw, R.C.; Jameson, V.J.; Walters, L.M.; Lafferty, M.H.; Powers, B.E.; Withrow, S.J. Evaluation of single-agent chemotherapy for treatment of clinically evident osteosarcoma metastases in dogs: 45 cases (1987-1991). J. Am. Vet. Med. Assoc. 1993, 202, 304–306. [Google Scholar] [PubMed]
- Dow, S.; Kurihara, J.; Regan, D. Cationic Lipid-DNA complexes induced spontaneous regression of adult-onset papillomatosis in dogs. 2015; in preparation. [Google Scholar]
- Veir, J.K.; Lappin, M.R.; Dow, S.W. Evaluation of a novel immunotherapy for treatment of chronic rhinitis in cats. J. Feline Med. Surg. 2006, 8, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-gamma in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, R.; Ten Hagen, T.L.; Eggermont, A.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Helfand, S.C.; Modiano, J.F.; Nowell, P.C. Immunophysiological studies of interleukin-2 and canine lymphocytes. Vet. Immunol. Immunopathol. 1992, 33, 1–16. [Google Scholar] [CrossRef]
- Helfand, S.C.; Soergel, S.A.; MacWilliams, P.S.; Hank, J.A.; Sondel, P.M. Clinical and immunological effects of human recombinant interleukin-2 given by repetitive weekly infusion to normal dogs. Cancer Immunol. Immunother. 1994, 39, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Helfand, S.C.; Soergel, S.A.; Modiano, J.F.; Hank, J.A.; Sondel, P.M. Induction of lymphokine-activated killer (LAK) activity in canine lymphocytes with low dose human recombinant interleukin-2 in vitro. Cancer Biother. 1994, 9, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Anderson, P.M.; Hasz, D.E.; Katsanis, E.; Neville, M.; Klausner, J.S. Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases. Cancer 1997, 79, 1409–1421. [Google Scholar] [CrossRef]
- Khanna, C.; Hasz, D.E.; Klausner, J.S.; Anderson, P.M. Aerosol delivery of interleukin 2 liposomes is nontoxic and biologically effective: Canine studies. Clin. Cancer Res. 1996, 2, 721–734. [Google Scholar] [PubMed]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, N.; Sanders, A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 1994, 174, 83–93. [Google Scholar] [CrossRef]
- Guth, A.M.; Hafeman, S.D.; Dow, S.W. Depletion of phagocytic myeloid cells triggers spontaneous T cell- and NK cell-dependent antitumor activity. Oncoimmunology 2012, 1, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Hafeman, S.; London, C.; Elmslie, R.; Dow, S. Evaluation of liposomal clodronate for treatment of malignant histiocytosis in dogs. Cancer Immunol. Immunother. 2010, 59, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Guth, A.M.; Hafeman, S.D.; Elmslie, R.E.; Dow, S.W. Liposomal clodronate treatment for tumour macrophage depletion in dogs with soft-tissue sarcoma. Vet. Comp. Oncol. 2013, 11, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T. Rational combinations of immunotherapeutics that target discrete pathways. J. Immunother. Cancer 2013, 1. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regan, D.; Dow, S. Manipulation of Innate Immunity for Cancer Therapy in Dogs. Vet. Sci. 2015, 2, 423-439. https://doi.org/10.3390/vetsci2040423
Regan D, Dow S. Manipulation of Innate Immunity for Cancer Therapy in Dogs. Veterinary Sciences. 2015; 2(4):423-439. https://doi.org/10.3390/vetsci2040423
Chicago/Turabian StyleRegan, Daniel, and Steven Dow. 2015. "Manipulation of Innate Immunity for Cancer Therapy in Dogs" Veterinary Sciences 2, no. 4: 423-439. https://doi.org/10.3390/vetsci2040423
APA StyleRegan, D., & Dow, S. (2015). Manipulation of Innate Immunity for Cancer Therapy in Dogs. Veterinary Sciences, 2(4), 423-439. https://doi.org/10.3390/vetsci2040423