Characterization of Global DNA Methylation in Different Gene Regions Reveals Candidate Biomarkers in Pigs with High and Low Levels of Boar Taint


Abstract
:1. Introduction
2. Materials and Methods
2.1. RRBS and Animal Data
2.2. Porcine Reference Gene Components
2.3. Differentially Methylated Gene Components, Significant Enrichments and Interaction Networks
3. Results
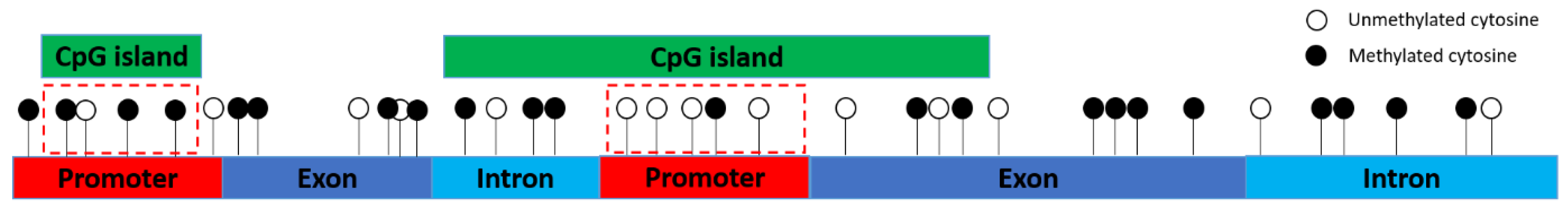
3.1. Methylation Levels of Promoters, Different Exons and Different Introns
3.2. Differentially Methylated Promoters, Exons, Introns and Their Overlaps with CpG Islands
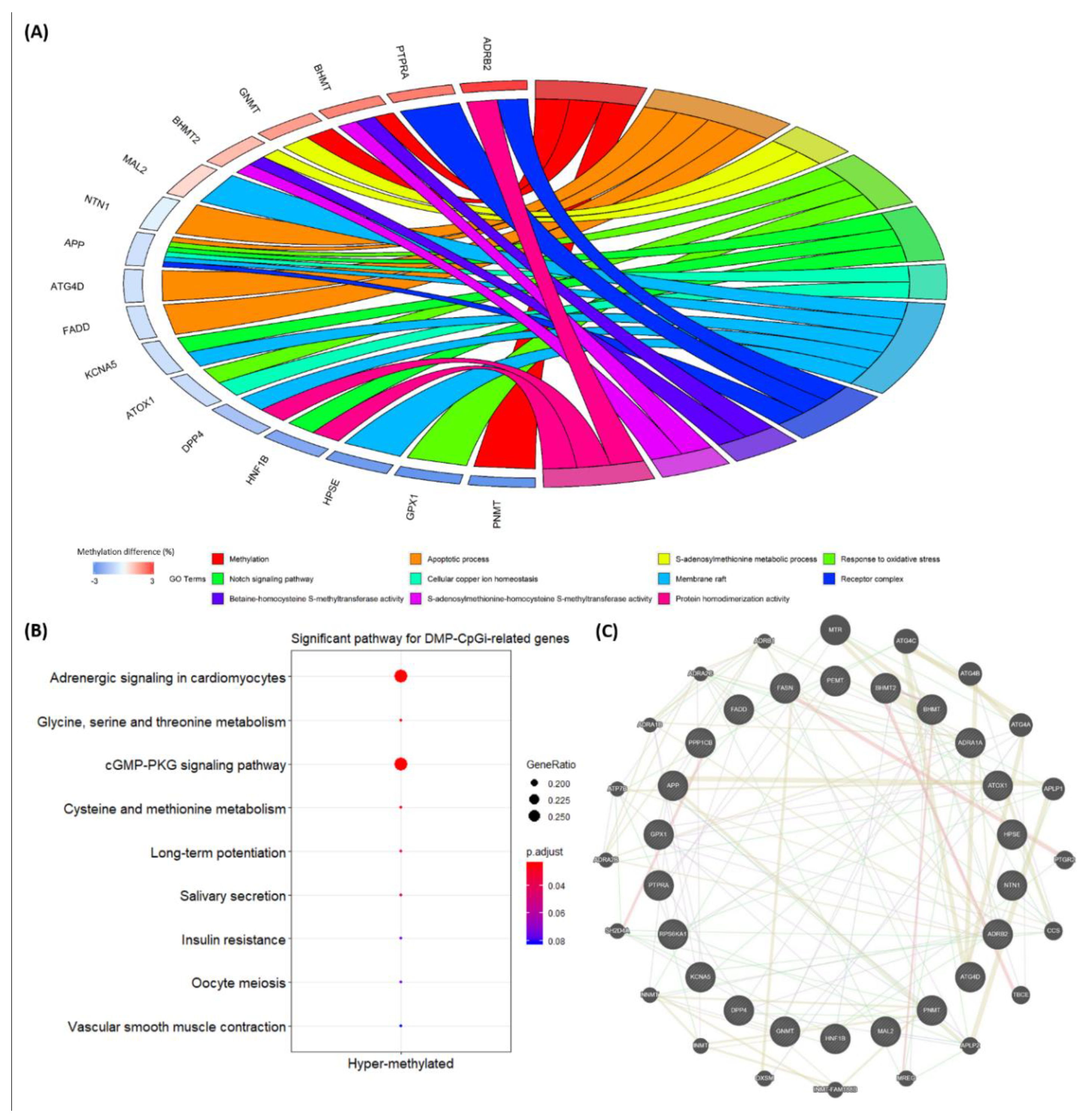
3.3. Biological Enrichment of DMP-CpGi-Related Genes and Interaction Networks
4. Discussion
4.1. Methylation Status of Promoters, Different Exons and Different Introns for BT
4.2. Biological Functions of DMP-CpGis-Related Genes
4.3. Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BT | Boar taint |
| CpG | Cytosine and guanine dinucleotide |
| DMC | Differentially methylated cytosine |
| DME | Differentially methylated exon |
| DME-CpGi | Differentially methylated exon in CpG island |
| DMI | Differentially methylated intron |
| DMI-CpGi | Differentially methylated intron in CpG island |
| DMP | Differentially methylated promoter |
| DMP-CpGi | Differentially methylated promoter in CpG island |
| DMR | Differentially methylated regions |
| EBV | Estimated breeding value |
| FDR | False discovery rate |
| GEO | Gene Expression Omnibus |
| HNS | Human nose score |
| RefSeqGene | Reference sequence gene |
| RRBS | Reduced representation bisulfite sequencing |
| SAM | S-adenosyl-methionine |
References
- Patterson, R.L.S. 5a-Androst-16-ene-3-one:-Compound responsible for boar taint in boar fat. J. Sci. Food Agric. 1968, 19, 31–38. [Google Scholar] [CrossRef]
- Gower, D.B. 16-Unsaturated C19steroids a review of their chemistry, biochemistry and possible physiological role. J. Steroid Biochem. 1972, 3, 45–103. [Google Scholar] [CrossRef]
- Bonneau, M.; Le Denmat, M.; Vaudelet, J.C.; Veloso Nunes, J.R.; Mortensen, A.B.; Mortensen, H.P. Contributions of fat androstenone and skatole to boar taint: I. Sensory attributes of fat and pork meat. Livest. Prod. Sci. 1992, 32, 63–80. [Google Scholar] [CrossRef]
- Malmfors, B.; Lundstrom, K. Consumer reactions to boar meat—A review. Livest. Prod. Sci. 1983, 10, 187–196. [Google Scholar] [CrossRef]
- Matthews, K.R.; Homer, D.B.; Punter, P.; Béague, M.P.; Gispert, M.; Kempster, A.J.; Agerhem, H.; Claudi-Magnussen, C.; Fischer, K.; Siret, F.; et al. An international study on the importance of androstenone and skatole for boar taint: III. Consumer survey in seven European countries. Meat Sci. 2000, 54, 271–283. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Hackett, J.A.; Reddington, J.P.; Nestor, C.E.; Dunican, D.S.; Branco, M.R.; Reichmann, J.; Reik, W.; Surani, M.A.; Adams, I.R.; Meehan, R.R. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 2012, 139, 3623–3632. [Google Scholar] [CrossRef] [Green Version]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 2006, 8, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 451, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Flores, K.; Wolschin, F.; Corneveaux, J.J.; Allen, A.N.; Huentelman, M.J.; Amdam, G.V. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genom. 2012, 13, 480. [Google Scholar] [CrossRef]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The importance of DNA methylation of exons on alternative splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, L.; Zhang, G. The association between DNA methylation and exon expression in the Pacific oyster Crassostrea gigas. PLoS ONE 2017, 12, e0185224. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, J.; Huang, S.; He, X. Genome-wide analysis reveals that exon methylation facilitates its selective usage in the human transcriptome. Brief. Bioinform. 2018, 19, 754–764. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 12, e14524. [Google Scholar] [CrossRef]
- Bieberstein, N.I.; Oesterreich, F.C.; Straube, K.; Neugebauer, K.M. First exon length controls active chromatin signatures and transcription. Cell Rep. 2012, 2, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Brinster, R.L.; Allen, J.M.; Behringer, R.R.; Gelinas, R.E.; Palmiter, R.D. Introns increase transcriptional efficiency in transgenic mice. Proc. Natl. Acad. Sci. USA 1988, 85, 836–840. [Google Scholar] [CrossRef] [Green Version]
- Callis, J.; Fromm, M.; Walbot, V. Introns increase gene expression in cultured maize cells. Genes Dev. 1987, 1, 1183–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furger, A.; O’Sullivan, J.M.; Binnie, A.; Lee, B.A.; Proudfoot, N.J. Promoter proximal splice sites enhance transcription. Genes Dev. 2002, 16, 2792–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, L.; Gianí, S.; Troina, F.; Breviario, D. Testing the IMEter on rice introns and other aspects of intron-mediated enhancement of gene expression. J. Exp. Bot. 2011, 62, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Kempe, K.; Rubtsova, M.; Riewe, D.; Gils, M. The production of male-sterile wheat plants through split barnase expression is promoted by the insertion of introns and flexible peptide linkers. Transgenic Res. 2013, 22, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shivakumar, M.; Han, S.; Sinclair, M.S.; Lee, Y.J.; Zheng, Y.; Olopade, O.I.; Kim, D.; Lee, Y. Population-dependent intron retention and DNA methylation in breast cancer. Mol. Cancer Res. 2018, 16, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Methylation at CpG islands in intron 1 of EGR2 confers enhancer-like activity. FEBS Lett. 2003, 554, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kadarmideen, H.N. Genome-wide DNA methylation analysis using next-generation sequencing to reveal candidate genes responsible for boar taint in pigs. Anim. Genet. 2019, 50, 644–659. [Google Scholar] [CrossRef]
- Wang, X.; Kadarmideen, H.N. An epigenome-wide DNA methylation map of testis in pigs for study of complex traits. Front. Genet. 2019, 10, 405. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Lee, J.; Le, M.T.; Nguyen, D.T.; Park, S.; Soundrarajan, N.; Schachtschneider, K.M.; Kim, J.; Park, J.K.; Kim, J.H.; et al. Genome-wide analysis of DNA methylation in pigs using reduced representation bisulfite sequencing. DNA Res. 2015, 22, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drag, M.; Skinkyté-Juskiené, R.; Do, D.N.; Kogelman, L.J.A.; Kadarmideen, H.N. Differential expression and co-expression gene networks reveal candidate biomarkers of boar taint in non-castrated pigs. Sci. Rep. 2017, 7, 12205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Franke, V.; Vlahoviček, K.; Mason, C.E.; Schübeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef]
- Wang, X.; Hao, D.; Kadarmideen, H.N. GeneDMRs: An R package for Gene-based Differentially Methylated Regions analysis. bioRxiv 2020, 04, 037168. [Google Scholar]
- Hochberg, B. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mostafavi, S.; Ray, D.; Warde-Farley, D.; Grouios, C.; Morris, Q. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008, 9, S4. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F.; Foret, S.; Kucharski, R.; Wolf, S.; Falckenhayn, C.; Maleszka, R. The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 2010, 8, e1000506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Shen, L.H.; Gui, L.X.; Yang, F.; Li, J.; Cao, S.Z.; Zuo, Z.C.; Ma, X.P.; Deng, J.L.; Ren, Z.H.; et al. Genome-wide DNA methylation profile of prepubertal porcine testis. Reprod. Fertil. Dev. 2018, 30, 349–358. [Google Scholar] [CrossRef]
- Chuang, T.J.; Chen, F.C.; Chen, Y.Z. Position-dependent correlations between DNA methylation and the evolutionary rates of mammalian coding exons. Proc. Natl. Acad. Sci. USA 2012, 109, 15841–15846. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Wang, X.; Li, Q.; Lian, J.; Li, L.; Jin, L.; Cai, H.; Xu, F.; Qi, H.; Zhang, L.; Wu, F.; et al. Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genom. 2014, 15, 1119. [Google Scholar] [CrossRef] [Green Version]
- Ganu, R.S.; Garrow, T.A.; Sodhi, M.; Rund, L.A.; Schook, L.B. Molecular characterization and analysis of the porcine betaine homocysteine methyltransferase and betaine homocysteine methyltransferase-2 genes. Gene 2011, 473, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Reyes, C.V.; Garrow, T.A. High sodium chloride intake decreases betaine-homocysteine S-methyltransferase expression in guinea pig liver and kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R182–R187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, F.; Miura, M. Enhancing S-adenosyl-methionine catabolism extends Drosophila lifespan. Nat. Commun. 2015, 6, 8332. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.N.; Baden, J.M.; Mathers, L.H.; Siddall, B.J.; Wong, D.L. Expression of phenylethanolamine N-methyltransferase in the embryonic rat heart. J. Mol. Cell. Cardiol. 1996, 28, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Fang, X.; Li, C.; Chen, C.; Liang, G.; Zheng, X.; Fan, Q. Increased PTPRA expression leads to poor prognosis through c-Src activation and G1 phase progression in squamous cell lung cancer. Int. J. Oncol. 2017, 51, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, S.; Gunawan, A.; Tholen, E.; Große-Brinkhaus, C.; Tesfaye, D.; Schellander, K.; Hofmann-Apitius, M.; Cinar, M.U.; Uddin, M.J. Pathway based analysis of genes and interactions influencing porcine testis samples from boars with divergent androstenone content in back fat. PLoS ONE 2014, 9, e91077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strathe, A.B.; Velander, I.H.; Mark, T.; Kadarmideen, H.N. Genetic parameters for androstenone and skatole as indicators of boar taint and their relationship to production and litter size traits in Danish Landrace. J. Anim. Sci. 2013, 91, 2587–2595. [Google Scholar] [CrossRef] [Green Version]
- Strathe, A.B.; Velander, I.H.; Mark, T.; Ostersen, T.; Hansen, C.; Kadarmideen, H.N. Genetic parameters for male fertility and its relationship to skatole and androstenone in Danish Landrace boars. J. Anim. Sci. 2013, 91, 4659–4668. [Google Scholar] [CrossRef] [Green Version]
- Claus, R.; Weiler, U.; Herzog, A. Physiological aspects of androstenone and skatole formation in the boar-A review with experimental data. Meat Sci. 1994, 38, 289–305. [Google Scholar] [CrossRef]
- Bonneau, M. Use of entire males for pig meat in the European Union. Meat Sci. 1998, 49, S257–S272. [Google Scholar] [CrossRef]
- Suravajhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omic data integration and analysis using systems genomics approaches: Methods and applications In animal production, health and welfare. Genet. Sel. Evol. 2016, 48, 38. [Google Scholar] [CrossRef] [Green Version]
- Kadarmideen, H.N. Genomics to systems biology in animal and veterinary sciences: Progress, lessons and opportunities. Livest. Sci. 2014, 166, 232–248. [Google Scholar] [CrossRef] [Green Version]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.J.; Karacaören, B.; de Koning, D.-J.; Lukic, B.; Hastings-Clark, N.; Velander, I.; Haley, C.S.; Archibald, A.L. Analysis of the genetics of boar taint reveals both single SNPs and regional effects. BMC Genom. 2014, 15, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, M.; Lien, S.; Aasmundstad, T.; Meuwissen, T.H.E.; Hansen, M.H.S.; Bendixen, C.; Grindflek, E. Association between SNPs within candidate genes and compounds related to boar taint and reproduction. BMC Genet. 2009, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Duijvesteijn, N.; Knol, E.F.; Merks, J.W.M.; Crooijmans, R.P.M.A.; Groenen, M.A.M.; Bovenhuis, H.; Harlizius, B. A genome-wide association study on androstenone levels in pigs reveals a cluster of candidate genes on chromosome 6. BMC Genet. 2010, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- de Campos, C.F.; Lopes, M.S.; de Silva, F.F.; Veroneze, R.; Knol, E.F.; Sávio Lopes, P.; Guimarães, S.E.F. Genomic selection for boar taint compounds and carcass traits in a commercial pig population. Livest. Sci. 2015, 174, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, A.; Sahadevan, S.; Cinar, M.U.; Neuhoff, C.; Große-Brinkhaus, C.; Frieden, L.; Tesfaye, D.; Tholen, E.; Looft, C.; Wondim, D.S.; et al. Identification of the novel candidate genes and variants in boar liver tissues with divergent skatole levels using RNA deep sequencing. PLoS ONE 2013, 11, e72298. [Google Scholar] [CrossRef] [Green Version]
- Drag, M.; Hansen, M.B.; Kadarmideen, H.N. Systems genomics study reveals expression quantitative trait loci, regulator genes and pathways associated with boar taint in pigs. PLoS ONE 2018, 13, e0192673. [Google Scholar] [CrossRef]
- Drag, M.; Kogelman, L.J.A.; Maribo, H.; Meinert, L.; Thomsen, P.D.; Kadarmideen, H.N. Charaterization of eQTLs associated with androstenone by RNA seuqecing in porcine testis. Physiol. Genom. 2019, 51, 488–499. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome | Gene Description | Region (Q-Value & Methylation Difference) | |||||
|---|---|---|---|---|---|---|---|---|
| TSPAN9 | 5 | Tetraspanin 9 | DMP (1.4 × 10−2 & 1.2%) | 1st DME (1.5 × 10−2 & −8.0%) | 6th DMI (1.7 × 10−10 & −5.5%) | DMP-CpGi (1.8 × 10−2 & 1.2%) | 1st DME-CpGi (1.9 × 10−2 & −8.0%) | 6th DMI-CpGi (2.5 × 10−5 & −12.8%) |
| MASP2 | 6 | Mannan binding lectin serine peptidase 2 | DMP (4.7 × 10−8 & −6.3%) | 10th DME (4.4 × 10−2 & −5.1%) | 9th DMI (5.8 × 10−5 & −8.5%) | NA | NA | NA |
| GPX1 | 13 | Glutathione peroxidase 1 | DMP (6.5 × 10−14 & −4.0%) | 2nd DME (2.5 × 10−2 & −2.1%) | 1st DMI (2.0 × 10−11 & −5.6%) | DMP-CpGi (6.6 × 10−14 & −4.0%) | 2nd DME-CpGi (3.1 × 10−2 & −2.1%) | 1st DMI-CpGi (2.1 × 10−11 & −5.6%) |
| Gene | Chromosome | Gene Description | Overlap Region | Q-Value | Methylation Difference (%) |
|---|---|---|---|---|---|
| POU2AF1 | 9 | POU class 2 homeobox- associating factor 1 | DMP-CpGi | 2.8 × 10−16 | −18.4 |
| IGFBP1 | 18 | Insulin-like growth factor-binding protein 1 | DMP-CpGi | 5.7 × 10−16 | 9.1 |
| GPX1 | 13 | Glutathione peroxidase 1 | DMP-CpGi | 6.6 × 10−14 | −4.0 |
| AMZ1 | 3 | Archaelysin family metallopeptidase 1 | DMP-CpGi | 2.1 × 10−11 | 7.9 |
| SLC7A14 | 13 | Solute carrier family 7 member 14 | DMP-CpGi | 5.2 × 10−11 | 2.1 |
| HOXA5 | 18 | Homeobox A5 | DMP-CpGi | 1.7 × 10−9 | 3.4 |
| PTPRA | 17 | Protein tyrosine phosphatase receptor type A | DMP-CpGi | 4.2 × 10−9 | 2.2 |
| PNMT | 12 | Phenylethanolamine N-methyltransferase | DMP-CpGi | 5.9 × 10−8 | −6.2 |
| PRM2 | 3 | Protamine 2 | DMP-CpGi | 1.1 × 10−7 | 14.0 |
| SOD3 | 8 | Superoxide dismutase 3 | DMP-CpGi | 2.5 × 10−7 | 4.7 |
| DCT | 11 | Dopachrome tautomerase | DMP-CpGi | 7.8 × 10−7 | 8.8 |
| CLEC4G | 2 | C-type lectin domain family 4 member G | DMP-CpGi | 3.9 × 10−6 | 11.9 |
| MIR671 | 18 | MicroRNA mir-671 | DMP-CpGi | 8.0 × 10−6 | −3.5 |
| TDRD10 | 4 | Tudor domain containing 10 | DMP-CpGi | 1.1 × 10−5 | 0.9 |
| LOC100519311 | 5 | Uncharacterized LOC100519311 | DMP-CpGi | 1.6 × 10−5 | 4.9 |
| CDH5 | 6 | Cadherin 5 | DMP-CpGi | 3.3 × 10−5 | −5.2 |
| BHMT | 2 | Betaine--homocysteine S-Methyltransferase | DMP-CpGi | 3.7 × 10−5 | 2.1 |
| TCTEX1D4 | 6 | Tctex1 domain containing 4 | DMP-CpGi | 7.4 × 10−5 | −6.1 |
| OXT | 17 | Oxytocin/neurophysin I prepropeptide | DMP-CpGi | 1.6 × 10−4 | −5.6 |
| ADRB2 | 2 | Adrenoceptor beta 2 | DMP-CpGi | 1.9 × 10−4 | 3.3 |
| ZNF217 | 17 | Zinc finger protein 217 | 3rd DME-CpGi | 8.0 × 10−32 | −8.2 |
| AMZ1 | 3 | Archaelysin family Metallopeptidase 1 | 7th DME-CpGi | 2.7 × 10−30 | −7.6 |
| YDJC | 14 | YdjC chitooligosaccharide Deacetylase homolog | 5th DME-CpGi | 6.9 × 10−24 | 24.2 |
| CHRM1 | 2 | Cholinergic receptor muscarinic 1 | 5th DME-CpGi | 8.8 × 10−22 | −8.2 |
| GLI2 | 15 | GLI family zinc finger 2 | 1st DME-CpGi | 5.2 × 10−21 | −10.4 |
| LRP8 | 6 | LDL receptor related protein 8 | 5th DME-CpGi | 1.6 × 10−20 | −13.9 |
| TNXB | 7 | Tenascin XB | 48th DME-CpGi | 6.4 × 10−17 | 4.3 |
| IGFBP1 | 18 | Insulin like growth factor binding protein 1 | 4th DME-CpGi | 5.7 × 10−16 | 9.1 |
| APOE | 6 | Apolipoprotein E | 4th DME-CpGi | 7.2 × 10−15 | −12.7 |
| CAPN2 | 10 | Calpain 2 | 6th DME-CpGi | 1.1 × 10−14 | −11.8 |
| FOXO3 | 1 | Forkhead box O3 | 2nd DME-CpGi | 1.5 × 10−14 | −9.1 |
| RTL1 | 7 | Retrotransposon Gag like 1 | 1st DME-CpGi | 2.3 × 10−14 | −4.2 |
| ADRA1D | 17 | Adrenoceptor alpha 1D | 3rd DME-CpGi | 7.5 × 10−14 | 4.4 |
| SIGIRR | 2 | Single Ig and TIR domain containing | 3rd DME-CpGi | 2.7 × 10−13 | 11.7 |
| ZNF205 | 3 | Zinc finger protein 205 | 1st DME-CpGi | 6.6 × 10−12 | 9.8 |
| SOX9 | 12 | SRY-box transcription factor 9 | 1st DME-CpGi | 5.4 × 10−10 | 2.9 |
| HOXA5 | 18 | Homeobox A5 | 1st DME-CpGi | 5.5 × 10−10 | 3.2 |
| COX10 | 12 | COX10 homolog, cytochrome c oxidase assembly protein, heme A: farnesyltransferase (yeast) | 7th DME-CpGi | 8.5 × 10−10 | −9.8 |
| KLF3 | 8 | Kruppel like factor 3 | 3rd DME-CpGi | 1.2 × 10−9 | −9.4 |
| MYO7A | 9 | Myosin VIIA | 16th DME-CpGi | 1.9 × 10−9 | −10.9 |
| CRYL1 | 11 | Crystallin lambda 1 | 6th DMI-CpGi | 2.7 × 10−22 | −7.6 |
| AUTS2 | 3 | Activator of transcription and developmental regulator AUTS2 | 5th DMI-CpGi | 5.2 × 10−21 | 3.9 |
| SCD5 | 8 | stearoyl-CoA desaturase 5 | 1st DMI-CpGi | 2.7 × 10−17 | −9.8 |
| SREBF1 | 12 | Sterol regulatory element binding transcription factor 1 | 18th DMI-CpGi | 1.5 × 10−14 | −5.1 |
| ABO | 1 | ABO, alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase | 2nd DMI-CpGi | 6.6 × 10−14 | −4.7 |
| BANP | 6 | BTG3 associated nuclear protein | 13th DMI-CpGi | 3.0 × 10−13 | 4.4 |
| PEMT | 12 | Phosphatidylethanolamine N-methyltransferase | 2nd DMI-CpGi | 3.6 × 10−13 | 8.7 |
| CTSD | 2 | Cathepsin D | 5th DMI-CpGi | 3.6 × 10−13 | −9.5 |
| TBCD | 12 | Tubulin folding cofactor D | 25th DMI-CpGi | 2.0 × 10−12 | −10.0 |
| GPX1 | 13 | Glutathione peroxidase 1 | 1st DMI-CpGi | 2.1 × 10−11 | −5.6 |
| SLC7A14 | 13 | Solute carrier family 7 member 14 | 7th DMI-CpGi | 5.2 × 10−11 | 2.1 |
| PDX1 | 11 | Pancreatic and duodenal homeobox 1 | 1st DMI-CpGi | 2.9 × 10−10 | −10.5 |
| PKD1 | 3 | Polycystin 1, transient receptor potential channel interacting | 1st DMI-CpGi | 3.3 × 10−10 | −8.0 |
| WDR45B | 12 | WD repeat domain 45B | 3rd DMI-CpGi | 6.2 × 10−10 | −12.2 |
| TBCD | 12 | Tubulin folding cofactor D | 24th DMI-CpGi | 1.8 × 10−9 | −20.9 |
| NOS3 | 18 | Nitric oxide synthase 3 | 2nd DMI-CpGi | 2.0 × 10−9 | 9.4 |
| TBCD | 12 | Tubulin folding cofactor D | 1st DMI-CpGi | 2.3 × 10−9 | −9.3 |
| BCO1 | 6 | Beta-carotene oxygenase 1 | 5th DMI-CpGi | 9.0 × 10−9 | −6.4 |
| HOXA10 | 18 | Homeobox A10 | 1st DMI-CpGi | 1.1 × 10−8 | 2.4 |
| TBCD | 12 | Tubulin folding cofactor D | 34th DMI-CpGi | 2.4 × 10−7 | 9.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Kadarmideen, H.N. Characterization of Global DNA Methylation in Different Gene Regions Reveals Candidate Biomarkers in Pigs with High and Low Levels of Boar Taint. Vet. Sci. 2020, 7, 77. https://doi.org/10.3390/vetsci7020077
Wang X, Kadarmideen HN. Characterization of Global DNA Methylation in Different Gene Regions Reveals Candidate Biomarkers in Pigs with High and Low Levels of Boar Taint. Veterinary Sciences. 2020; 7(2):77. https://doi.org/10.3390/vetsci7020077
Chicago/Turabian StyleWang, Xiao, and Haja N. Kadarmideen. 2020. "Characterization of Global DNA Methylation in Different Gene Regions Reveals Candidate Biomarkers in Pigs with High and Low Levels of Boar Taint" Veterinary Sciences 7, no. 2: 77. https://doi.org/10.3390/vetsci7020077