Effects of ACTH-Induced Long-Term Hypercortisolism on the Transcriptome of Canine Visceral Adipose Tissue

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments and Tissue Sampling
2.2. RNA Extraction
2.3. RNA Sequencing (RNA-Seq)
2.4. Data Analysis
3. Results
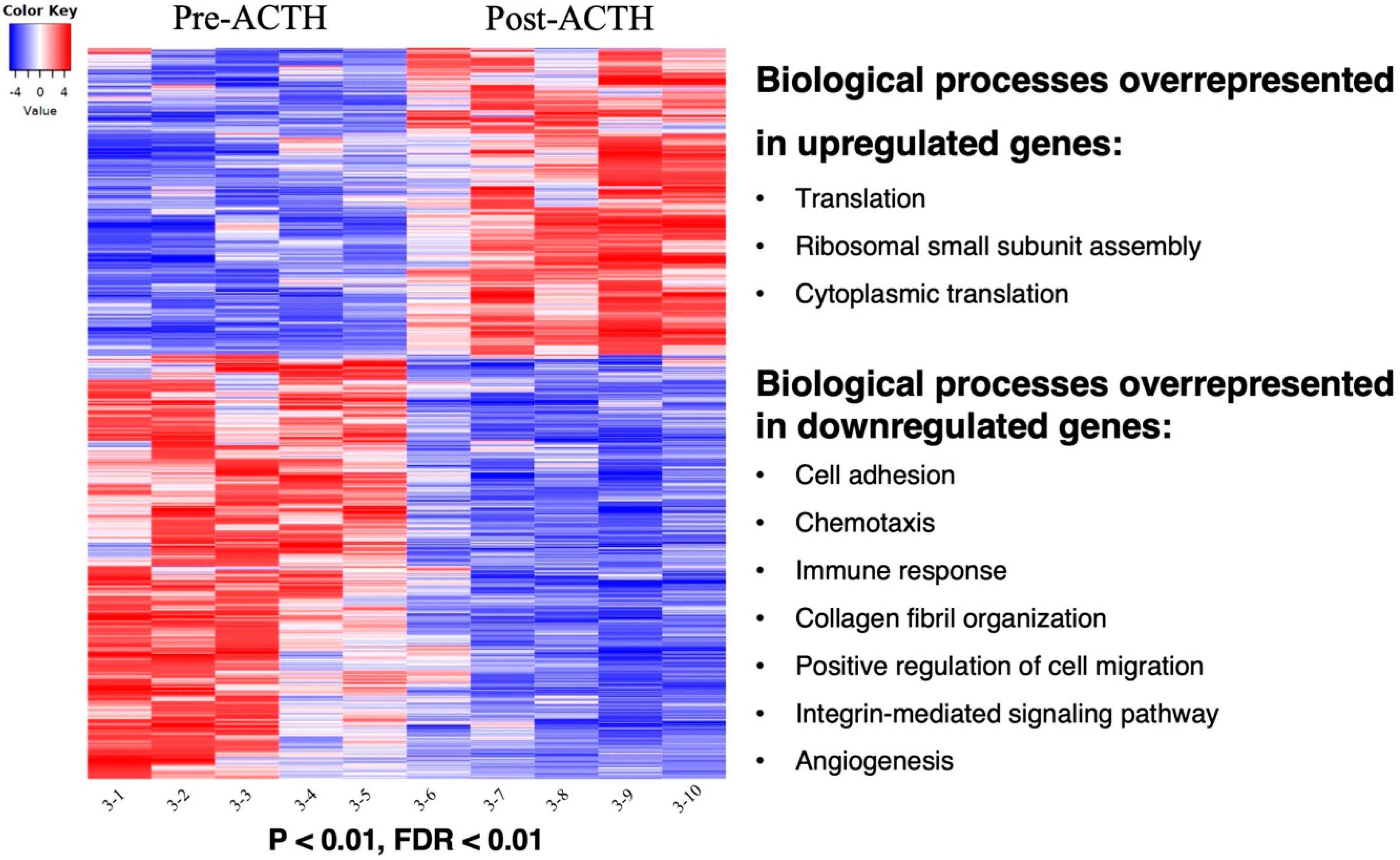
3.1. Quality Control and Pairwise Comparison
3.2. Functional Annotations
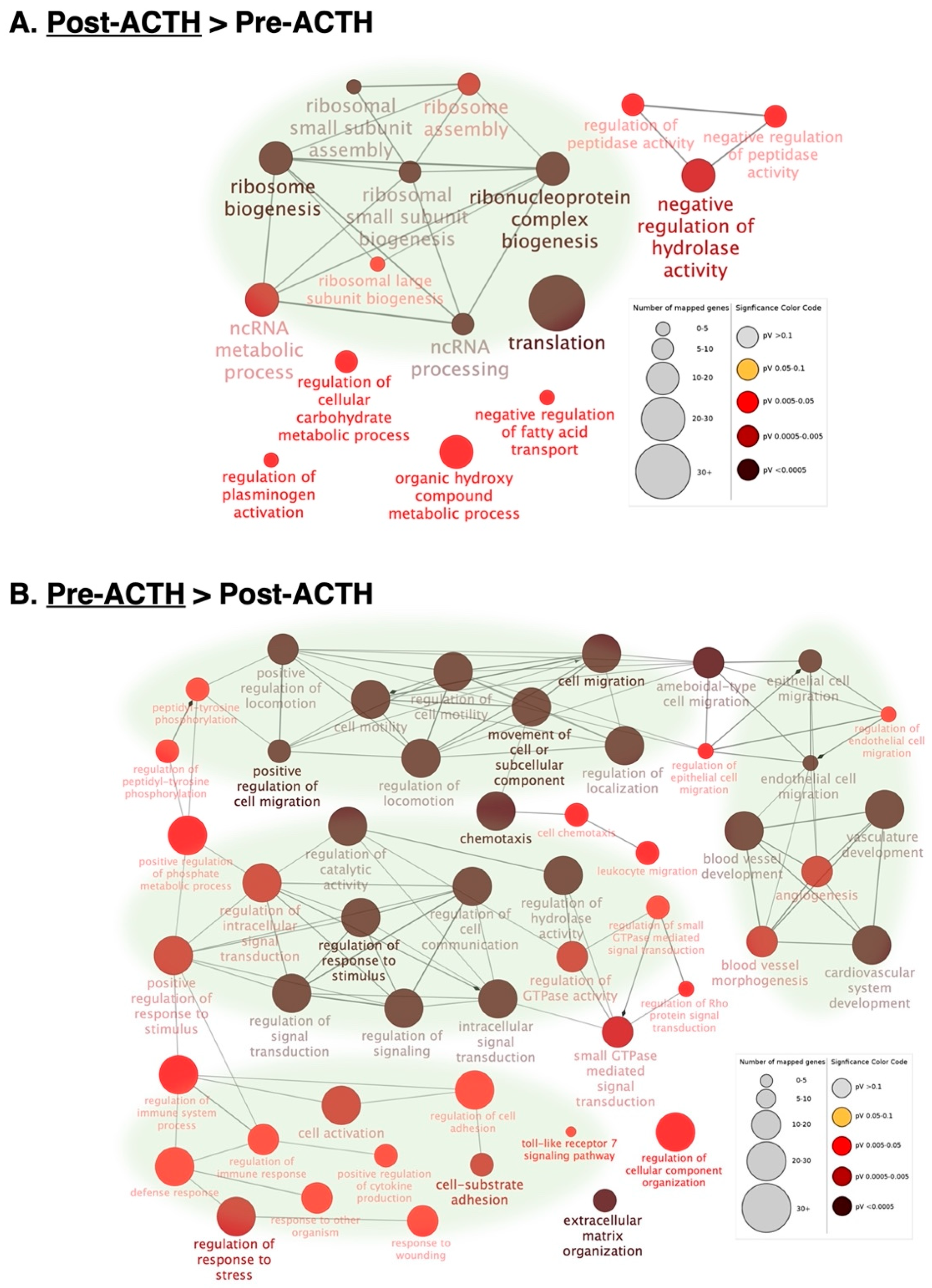
3.2.1. Gene Ontologies
3.2.2. Functional Networks
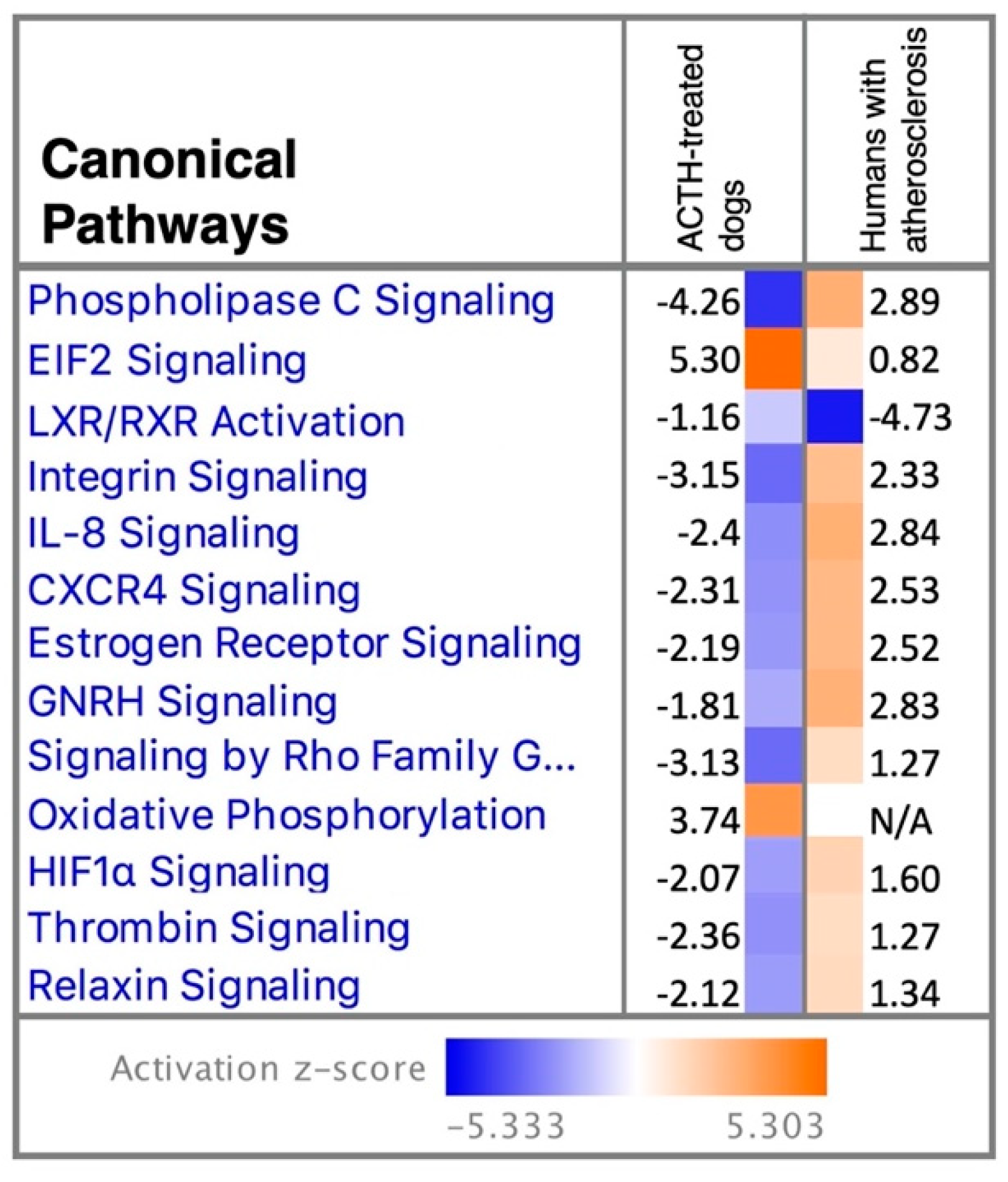
3.2.3. Canonical Pathways and Upstream Regulators
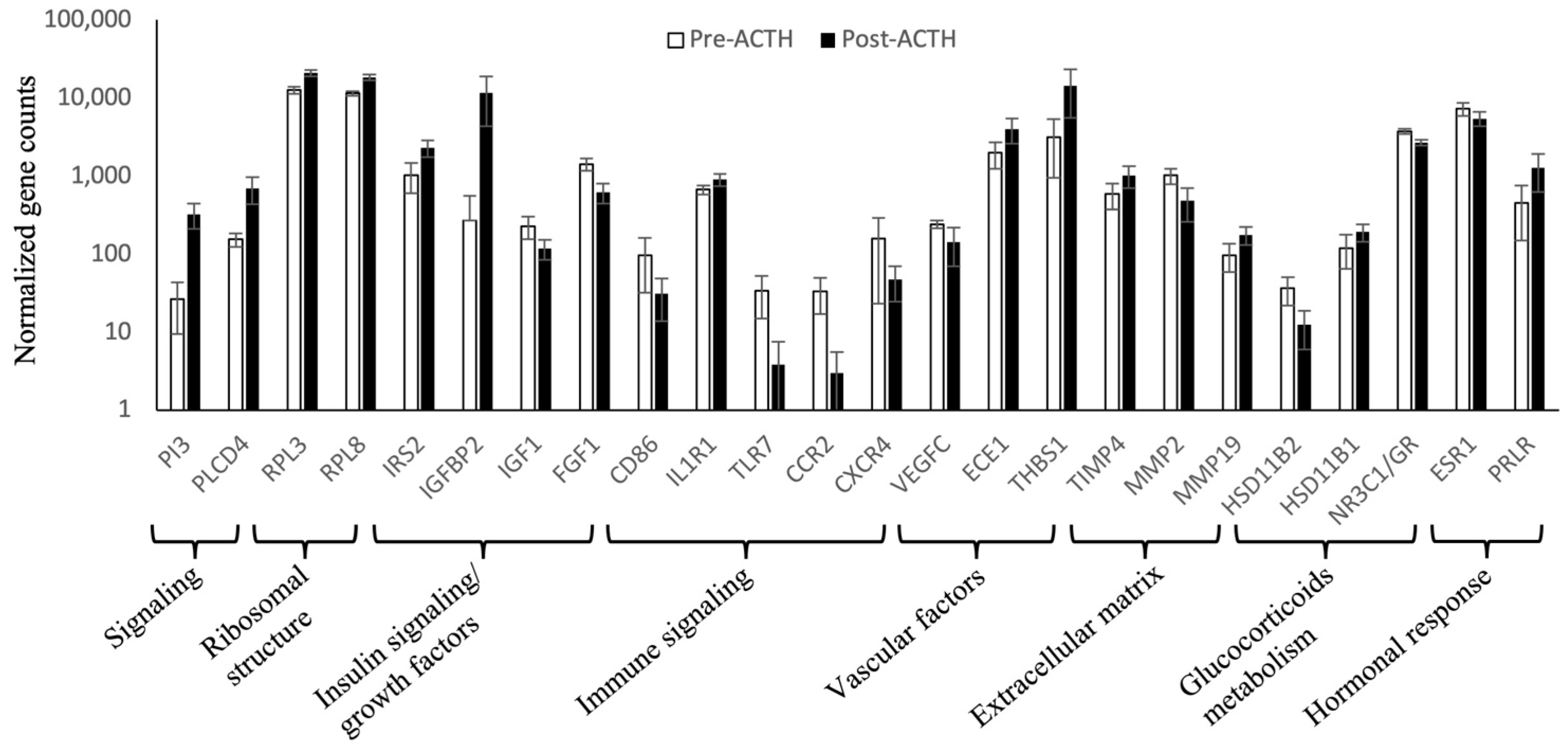
3.3. Normalized Counts of Representative Genes
3.4. Analysis Match
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Behrend, E.N.; Kooistra, H.S.; Nelson, R.; Reusch, C.E.; Scott-Moncrieff, J.C. Diagnosis of spontaneous canine hyperadrenocorticism: 2012 ACVIM consensus statement (small animal). J. Vet. Intern. Med. 2013, 27, 1292–1304. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid Res. 2012, 53, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, R.S.; KAss, P.H.; Van Winkle, T.J. Association between Diabetes Mellitus, Hypothyroidism or Hyperadrenocorticism, and Atherosclerosis in Dogs. J. Vet. Intern. Med. 2003, 17, 489–494. [Google Scholar] [CrossRef]
- Hess, R.S. Insulin resistance in dogs. Vet. Clin. Small Anim. Pract. 2010, 40, 309–316. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schurmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [Green Version]
- Sieber-Ruckstuhl, N.S.; Meli, M.L.; Boretti, F.S.; Gonczi, E.; Lutz, H.; Reusch, C.E. Quantitative real-time PCR for the measurement of 11beta-HSD1 and 11beta-HSD2 mRNA levels in tissues of healthy dogs. Horm. Metab. Res. 2007, 39, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Chau, Y.Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Leray, V.; Serisier, S.; Khosniat, S.; Martin, L.; Dumon, H.; Nguyen, P. Adipose tissue gene expression in obese dogs after weight loss. J. Anim. Physiol. Anim. Nutr. 2008, 92, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus white adipose tissue: A mini-review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Raff, H.; Carroll, T. Cushing’s syndrome: From physiological principles to diagnosis and clinical care. J. Physiol. 2015, 593, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrau, F.; Korbonits, M. Metabolic comorbidities in Cushing’s syndrome. Eur. J. Endocrinol. 2015, 173, M133–M157. [Google Scholar] [CrossRef] [Green Version]
- Simonyte, K.; Rask, E.; Naslund, I.; Angelhed, J.E.; Lonn, L.; Olsson, T.; Mattsson, C. Obesity is accompanied by disturbances in peripheral glucocorticoid metabolism and changes in FA recycling. Obesity 2009, 17, 1982–1987. [Google Scholar] [CrossRef]
- Krsek, M.; Rosická, M.; Nedvídková, J.; Kvasnicková, H.; Hána, V.; Marek, J.; Haluzík, M.; Lai, E.W.; Pacák, K. Increased lipolysis of subcutaneous abdominal adipose tissue and altered noradrenergic activity in patients with Cushing’s syndrome: An in-vivo microdialysis study. Physiol. Res. 2006, 55, 421–428. [Google Scholar] [CrossRef]
- Campbell, J.E.; Peckett, A.J.; D’Souza, A.M.; Hawke, T.J.; Riddell, M.C. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol. 2011, 300, C198–C209. [Google Scholar] [CrossRef] [Green Version]
- Dalle, H.; Garcia, M.; Antoine, B.; Boehm, V.; Do, T.T.H.; Buyse, M.; Ledent, T.; Lamaziere, A.; Magnan, C.; Postic, C.; et al. Adipocyte Glucocorticoid Receptor Deficiency Promotes Adipose Tissue Expandability and Improves the Metabolic Profile Under Corticosterone Exposure. Diabetes 2019, 68, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, R.; Okuno, Y.; Mukai, K.; Kitamura, T.; Hayakawa, T.; Onodera, T.; Murata, M.; Fukuhara, A.; Imamura, R.; Miyagawa, Y.; et al. Adipocyte GR Inhibits Healthy Adipose Expansion Through Multiple Mechanisms in Cushing Syndrome. Endocrinology 2019, 160, 504–521. [Google Scholar] [CrossRef] [Green Version]
- Sieber-Ruckstuhl, N.S.; Burla, B.; Spoerel, S.; Schmid, F.; Venzin, C.; Cazenave-Gassiot, A.; Bendt, A.K.; Torta, F.; Wenk, M.R.; Boretti, F.S. Changes in the Canine Plasma Lipidome after Short- and Long-Term Excess Glucocorticoid Exposure. Sci. Rep. 2019, 9, 6015. [Google Scholar] [CrossRef] [Green Version]
- Zatta, S.; Rehrauer, H.; Gram, A.; Boos, A.; Kowalewski, M.P. Transcriptome analysis reveals differences in mechanisms regulating cessation of luteal function in pregnant and non-pregnant dogs. BMC Genom. 2017, 18, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, M.; Opitz, L.; Russo, G.; Qi, W.; Schlapbach, R.; Rehrauer, H. SUSHI: An exquisite recipe for fully documented, reproducible and reusable NGS data analysis. BMC Bioinform. 2016, 17, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Schlapbach, R.; Rehrauer, H. RNA-Seq Data Analysis: From Raw Data Quality Control to Differential Expression Analysis. Methods Mol. Biol. 2017, 1669, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hagg, S.; Skogsberg, J.; Lundstrom, J.; Noori, P.; Nilsson, R.; Zhong, H.; Maleki, S.; Shang, M.M.; Brinne, B.; Bradshaw, M.; et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: The Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009, 5, e1000754. [Google Scholar] [CrossRef] [Green Version]
- Divertie, G.D.; Jensen, M.D.; Miles, J.M. Stimulation of Lipolysis in Humans by Physiological Hypercortisolemia. Diabetes 1991, 40, 1228–1232. [Google Scholar] [CrossRef]
- Hochberg, I.; Harvey, I.; Tran, Q.T.; Stephenson, E.J.; Barkan, A.L.; Saltiel, A.R.; Chandler, W.F.; Bridges, D. Gene expression changes in subcutaneous adipose tissue due to Cushing’s disease. J. Mol. Endocrinol. 2015, 55, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.M. Tissue-specific Cushing’s syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur. J. Endocrinol. 2003, 149, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Merrick, W.C.; Pavitt, G.D. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [Google Scholar] [CrossRef]
- Fonseca, B.D.; Lahr, R.M.; Damgaard, C.K.; Alain, T.; Berman, A.J. LARP1 on TOP of ribosome production. Wiley Interdiscip. Rev. RNA 2018, 9, e1480. [Google Scholar] [CrossRef]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.Q.; Phillips, C.T.; et al. A PPARgamma-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef]
- Fan, L.; Ding, L.; Lan, J.; Niu, J.; He, Y.; Song, L. Fibroblast Growth Factor-1 Improves Insulin Resistance via Repression of JNK-Mediated Inflammation. Front. Pharmacol. 2019, 10, 1478. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; van Dijk, T.H.; et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014, 513, 436–439. [Google Scholar] [CrossRef]
- Jaye, M.; Howk, R.; Burgess, W.; Ricca, G.A.; Chiu, I.M.; Ravera, M.W.; O’Brien, S.J.; Modi, W.S.; Maciag, T.; Drohan, W.N. Human endothelial cell growth factor: Cloning, nucleotide sequence, and chromosome localization. Science 1986, 233, 541–545. [Google Scholar] [CrossRef]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [CrossRef]
- Bornstein, P. Thrombospondins function as regulators of angiogenesis. J. Cell Commun. Signal. 2009, 3, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Kong, P.; Gonzalez-Quesada, C.; Li, N.; Cavalera, M.; Lee, D.W.; Frangogiannis, N.G. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E439–E450. [Google Scholar] [CrossRef] [Green Version]
- Mejia-Cristobal, L.M.; Reus, E.; Lizarraga, F.; Espinosa, M.; Ceballos-Cancino, G.; Lopez, T.V.; Garay, S.; Maldonado, V.; Melendez-Zajgla, J. Tissue inhibitor of metalloproteases-4 (TIMP-4) modulates adipocyte differentiation in vitro. Exp. Cell Res. 2015, 335, 207–215. [Google Scholar] [CrossRef]
- Sakamuri, S.; Watts, R.; Takawale, A.; Wang, X.; Hernandez-Anzaldo, S.; Bahitham, W.; Fernandez-Patron, C.; Lehner, R.; Kassiri, Z. Absence of Tissue Inhibitor of Metalloproteinase-4 (TIMP4) ameliorates high fat diet-induced obesity in mice due to defective lipid absorption. Sci. Rep. 2017, 7, 6210. [Google Scholar] [CrossRef]
- Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Armstrong, M.J.; Borrows, S.; Yu, J.; Wagenmakers, A.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoids fail to cause insulin resistance in human subcutaneous adipose tissue in vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Gathercole, L.L.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoid modulation of insulin signaling in human subcutaneous adipose tissue. J. Clin. Endocrinol. Metab. 2007, 92, 4332–4339. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, J.J.; Boudreau, A.; Wu, D.; Abdou Salem, H.; Carrigan, A.; Gagnon, A.; Mears, A.J.; Sorisky, A.; Atlas, E.; Hache, R.J. Insulin sensitization of human preadipocytes through glucocorticoid hormone induction of forkhead transcription factors. Mol. Endocrinol. 2010, 24, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Geer, E.B.; Islam, J.; Buettner, C. Mechanisms of glucocorticoid-induced insulin resistance: Focus on adipose tissue function and lipid metabolism. Endocrinol. Metab. Clin. N. Am. 2014, 43, 75–102. [Google Scholar] [CrossRef] [Green Version]
- Stimson, R.H.; Anderson, A.J.; Ramage, L.E.; Macfarlane, D.P.; de Beaux, A.C.; Mole, D.J.; Andrew, R.; Walker, B.R. Acute physiological effects of glucocorticoids on fuel metabolism in humans are permissive but not direct. Diabetes Obes. Metab. 2017, 19, 883–891. [Google Scholar] [CrossRef]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [Green Version]
- Kershaw, E.E.; Morton, N.M.; Dhillon, H.; Ramage, L.; Seckl, J.R.; Flier, J.S. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes 2005, 54, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Carre, N.; Binart, N. Prolactin and adipose tissue. Biochimie 2014, 97, 16–21. [Google Scholar] [CrossRef]
- Cvoro, A.; Yuan, C.; Paruthiyil, S.; Miller, O.H.; Yamamoto, K.R.; Leitman, D.C. Cross talk between glucocorticoid and estrogen receptors occurs at a subset of proinflammatory genes. J. Immunol. 2011, 186, 4354–4360. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Franchimont, D.; Hiroi, N.; Frey, G.; Boettner, A.; Ehrhart-Bornstein, M.; O’shea, J.J.; Chrousos, G.P.; Bornstein, S.R. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002, 16, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Hasenmajer, V.; Sbardella, E.; Sciarra, F.; Minnetti, M.; Isidori, A.M.; Venneri, M.A. The Immune System in Cushing’s Syndrome. Trends Endocrinol. Metab. 2020, 31, 655–669. [Google Scholar] [CrossRef]
- Kronfol, Z.; Starkman, M.; Schteingart, D.E.; Singh, V.; Zhang, Q.; Hill, E. Immune regulation in cushing’s syndrome: Relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996, 21, 599–608. [Google Scholar] [CrossRef]
- Lee, I.T.; Atuahene, A.; Egritag, H.E.; Wang, L.; Donovan, M.; Buettner, C.; Geer, E.B. Active Cushing Disease Is Characterized by Increased Adipose Tissue Macrophage Presence. J. Clin. Endocrinol. Metab. 2019, 104, 2453–2461. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Gencer, S.; Evans, B.R.; van der Vorst, E.P.C.; Doring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.-Q.; Huang, W.; Wang, Y. SDF-1α and CXCR4 as therapeutic targets in cardiovascular disease. Am. J. Cardiovasc. Dis. 2012, 2, 20–28. [Google Scholar]
- Neary, N.M.; Booker, O.J.; Abel, B.S.; Matta, J.R.; Muldoon, N.; Sinaii, N.; Pettigrew, R.I.; Nieman, L.K.; Gharib, A.M. Hypercortisolism is associated with increased coronary arterial atherosclerosis: Analysis of noninvasive coronary angiography using multidetector computerized tomography. J. Clin. Endocrinol. Metab. 2013, 98, 2045–2052. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavares Pereira, M.; Martin, I.; Rehrauer, H.; Kowalewski, M.P.; Boretti, F.S.; Sieber-Ruckstuhl, N.S. Effects of ACTH-Induced Long-Term Hypercortisolism on the Transcriptome of Canine Visceral Adipose Tissue. Vet. Sci. 2022, 9, 250. https://doi.org/10.3390/vetsci9060250
Tavares Pereira M, Martin I, Rehrauer H, Kowalewski MP, Boretti FS, Sieber-Ruckstuhl NS. Effects of ACTH-Induced Long-Term Hypercortisolism on the Transcriptome of Canine Visceral Adipose Tissue. Veterinary Sciences. 2022; 9(6):250. https://doi.org/10.3390/vetsci9060250
Chicago/Turabian StyleTavares Pereira, Miguel, Isabelle Martin, Hubert Rehrauer, Mariusz P. Kowalewski, Felicitas S. Boretti, and Nadja S. Sieber-Ruckstuhl. 2022. "Effects of ACTH-Induced Long-Term Hypercortisolism on the Transcriptome of Canine Visceral Adipose Tissue" Veterinary Sciences 9, no. 6: 250. https://doi.org/10.3390/vetsci9060250