
Ex Vivo Antiplatelet Effects of Oral Anticoagulants

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Methods
2.1. Blood Samples
2.2. Calibrated Automated Measurement of Thrombin Generation (CAT)
2.3. Platelet Aggregation Studies
2.4. Thromboxane Generation
2.5. Flow Cytometry
2.6. Statistical Analyses
3. Results
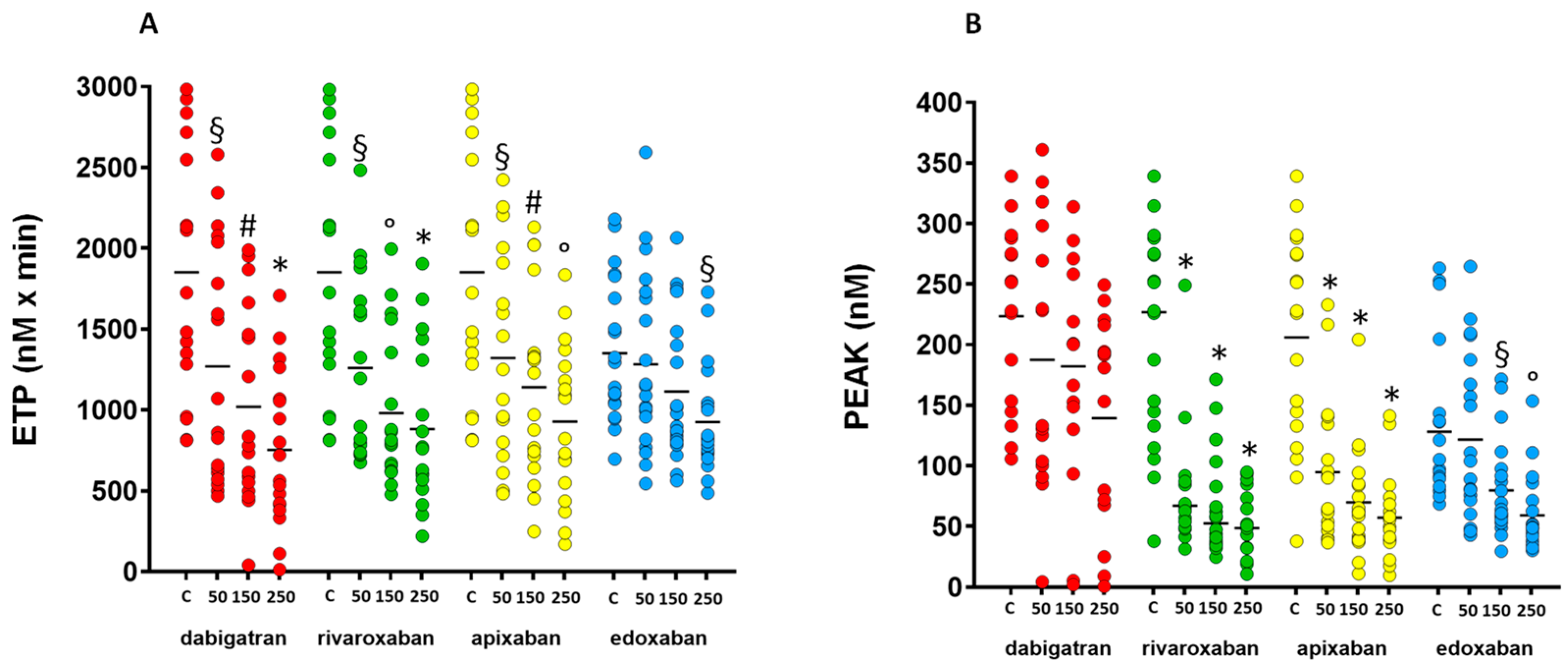
3.1. Calibrated Automated Measurement of Thrombin Generation
3.2. Platelet Aggregation
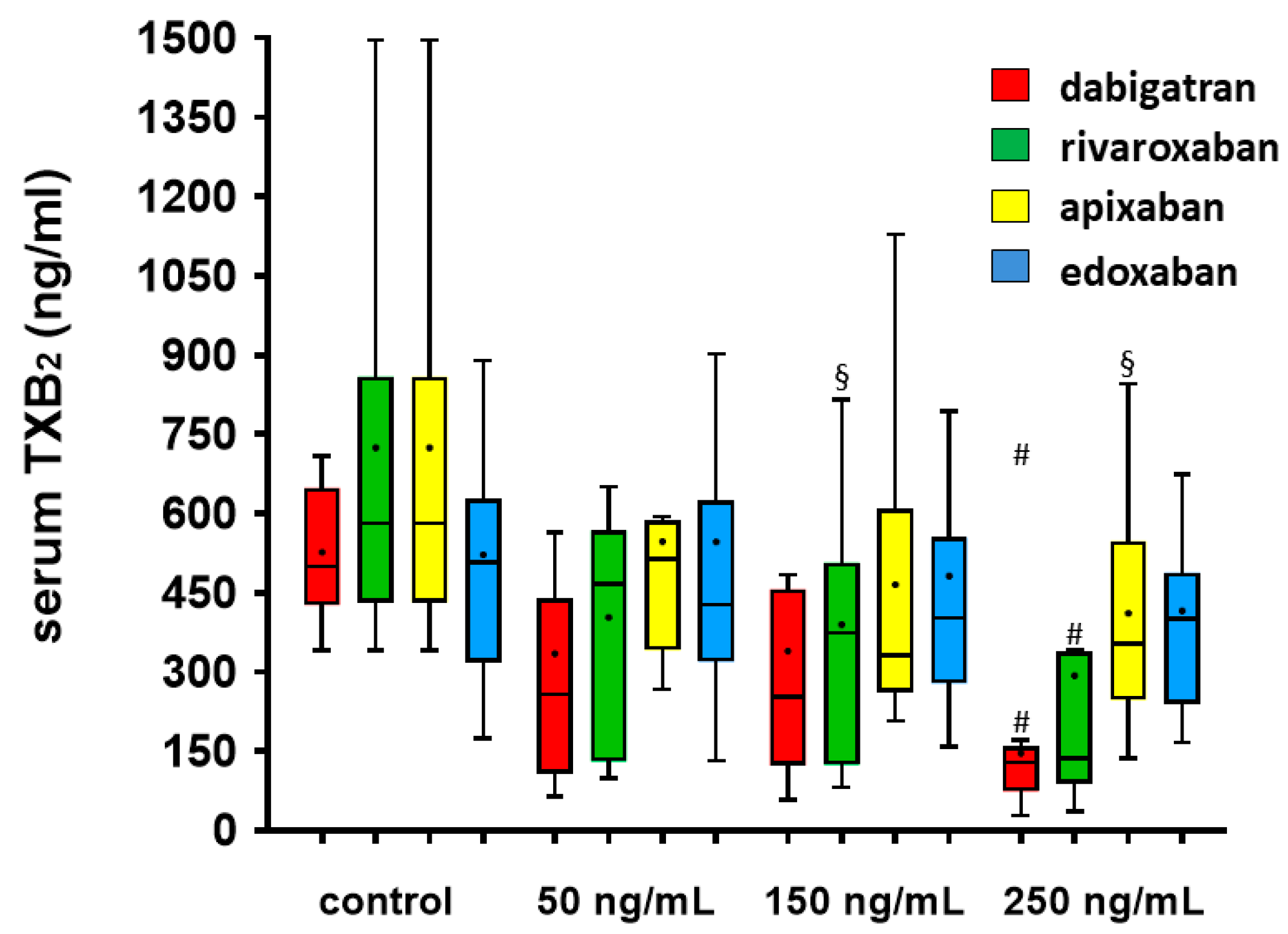
3.3. Serum TXB2 Generation
3.4. Flow Cytometry
4. Discussion
- All NOACs reduced thrombin generation measured by CAT; ETP is the parameter more consistently affected by the addition of NOACs at different concentrations;
- Platelet aggregation induced by ADP and TRAP was not affected by the addition of any NOACs; conversely, platelet aggregation induced by thrombin was significantly reduced by the addition of dabigatran, and TF-induced platelet aggregation is inhibited by FXa inhibitors;
- Serum TX generation was reduced by the addition of all NOACs;
- The expression of PAR-1 on the platelet surface, as evaluated by flow cytometry, was increased by the addition of dabigatran in a concentration-dependent manner, without an enhancement of platelet activation.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Wolzt, M.; Bostrom, S.L.; Svensson, M.; Wahlander, K.; Grind, M.; Sarich, T.C. Effects of the oral direct thrombin inhibitor ximelagatran on p-selectin expression and thrombin generation in atrial fibrillation. Pathophysiol. Haemost. Thromb. 2003, 33, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Mattsson, C. Thrombin-induced platelet activation and its inhibition by anticoagulants with different modes of action. Blood Coagul. Fibrinolysis 2003, 14, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, R.; Thom, J.; Wood, A.; Phillips, M.; Muhammad, S.; Baker, R. Thrombin generation using the calibrated automated thrombinoscope to assess reversibility of dabigatran and rivaroxaban. Thromb. Haemost. 2014, 111, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Heitmeier, S.; Laux, V. Effects of Rivaroxaban on Platelet Activation and Platelet-Coagulation Pathway Interaction: In Vitro and In Vivo Studies. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Padovan, L.; Veena, C.; Scalambrino, E.; Testa, S.; Peyvandi, F. How the direct oral anticoagulant apixaban affects thrombin generation parameters. Thromb. Res. 2015, 135, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Yang, Y.; Zhu, J.; Wu, S.; Zhou, Z.; Huang, B.; Wang, J.; Shao, X.; Zhang, H. An in-vitro evaluation of direct thrombin inhibitor and factor Xa inhibitor on tissue factor-induced thrombin generation and platelet aggregation: A comparison of dabigatran and rivaroxaban. Blood Coagul. Fibrinolysis 2016, 27, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Sokol, J.; Nehaj, F.; Ivankova, J.; Mokan, M.; Mokan, M.; Stasko, J. Dabigatran affects thrombin-dependent platelet aggregation after a week-long therapy. Scand. Cardiovasc. J. 2018, 52, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Sokol, J.; Nehaj, F.; Ivankova, J.; Mokan, M.; Mokan, M. First evidence: Rivaroxaban and apixaban reduce thrombin-dependent platelet aggregation. J. Thromb. Thrombolysis 2018, 46, 393–398. [Google Scholar] [CrossRef]
- Van Ryn, J.; Kink-Eiband, M.; Hauel, N.; Priepke, H.; Wienen, W. Effects of Dabigatran, a Direct Thrombin Inhibitor, as Compared to the Direct Factor Xa Inhibitors, Rivaroxaban and Apixaban, on Tissue Factor-Induced Human Platelet Aggregation in Platelet Rich Plasma. Blood 2007, 110, 1884. [Google Scholar] [CrossRef]
- Ringwala, S.M.; DiBattiste, P.M.; Schneider, D.J. Effects on platelet function of a direct acting antagonist of coagulation factor Xa. J. Thromb. Thrombolysis 2012, 34, 291–296. [Google Scholar] [CrossRef]
- Honda, Y.; Kamisato, C.; Morishima, Y. Edoxaban, a direct factor Xa inhibitor, suppresses tissue-factor induced human platelet aggregation and clot-bound factor Xa in vitro: Comparison with an antithrombin-dependent factor Xa inhibitor, fondaparinux. Thromb. Res. 2016, 141, 17–21. [Google Scholar] [CrossRef]
- Wong, P.C.; Jiang, X. Apixaban, a direct factor Xa inhibitor, inhibits tissue-factor induced human platelet aggregation in vitro: Comparison with direct inhibitors of factor VIIa, XIa and thrombin. Thromb. Haemost. 2010, 104, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Vinholt, P.J.; Nielsen, C.; Söderström, A.C.; Brandes, A.; Nybo, M. Dabigatran reduces thrombin-induced platelet aggregation and activation in a dose-dependent manner. J. Thromb. Thrombolysis 2017, 44, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Nehaj, F.; Sokol, J.; Mokan, M.; Ivankova, J.; Mokan, M. Thrombin Receptor Agonist Peptide-Induced Platelet Aggregation Is Reduced in Patients Receiving Dabigatran. Clin. Appl. Thromb. Hemost. 2018, 24, 268–272. [Google Scholar] [CrossRef]
- Olivier, C.B.; Weik, P.; Meyer, M.; Weber, S.; Diehl, P.; Bode, C.; Moser, M.; Zhou, Q. Dabigatran and rivaroxaban do not affect AA- and ADP-induced platelet aggregation in patients receiving concomitant platelet inhibitors. J. Thromb. Thrombolysis 2016, 42, 161–166. [Google Scholar] [CrossRef]
- Chen, B.; Soto, A.G.; Coronel, L.J.; Goss, A.; van Ryn, J.; Trejo, J. Characterization of thrombin-bound dabigatran effects on protease-activated receptor-1 expression and signaling in vitro. Mol. Pharmacol. 2015, 88, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Achilles, A.; Mohring, A.; Dannenberg, L.; Grandoch, M.; Hohlfeld, T.; Fischer, J.W.; Levkau, B.; Kelm, M.; Zeus, T.; Polzin, A. Dabigatran enhances platelet reactivity and platelet thrombin receptor expression in patients with atrial fibrillation. J. Thromb. Haemost. 2017, 15, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Steppich, B.; Dobler, F.; Brendel, L.C.; Hessling, G.; Braun, S.L.; Steinsiek, A.L.; Deisenhofer, I.; Hyseni, A.; Roest, M.; Ott, I. Effect of the FXa inhibitors Rivaroxaban and Apixaban on platelet activation in patients with atrial fibrillation. J. Thromb. Thrombolysis 2017, 43, 490–497. [Google Scholar] [CrossRef]
- Banovcin, P., Jr.; Skornova, I.; Samos, M.; Schnierer, M.; Bolek, T.; Kovar, F.; Stasko, J.; Kubisz, P.; Mokan, M. Platelet Aggregation in Direct Oral Factor Xa Inhibitors-treated Patients With Atrial Fibrillation: A Pilot Study. J. Cardiovasc. Pharmacol. 2017, 70, 263–266. [Google Scholar] [CrossRef]
- Nehaj, F.; Sokol, J.; Ivankova, J.; Mokan, M.; Kovar, F.; Stasko, J.; Mokan, M. First Evidence: TRAP-Induced Platelet Aggregation Is Reduced in Patients Receiving Xabans. Clin. Appl. Thromb./Hemost. 2018, 24, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Mueck, W.; Stampfuss, J.; Kubitza, D.; Becka, M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin. Pharmacokinet. 2014, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Parasrampuria, D.A.; Truitt, K.E. Pharmacokinetics and Pharmacodynamics of Edoxaban, a Non-Vitamin K Antagonist Oral Anticoagulant that Inhibits Clotting Factor Xa. Clin. Pharmacokinet. 2016, 55, 641–655. [Google Scholar] [CrossRef]
- Testa, S.; Tripodi, A.; Legnani, C.; Pengo, V.; Abbate, R.; Dellanoce, C.; Carraro, P.; Salomone, L.; Paniccia, R.; Paoletti, O.; et al. Plasma levels of direct oral anticoagulants in real life patients with atrial fibrillation: Results observed in four anticoagulation clinics. Thromb. Res. 2016, 137, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Dunois, C. Laboratory Monitoring of Direct Oral Anticoagulants (DOACs). Biomedicines 2021, 9, 445. [Google Scholar] [CrossRef]
- Perzborn, E.; Strassburger, J.; Wilmen, A.; Pohlmann, J.; Roehrig, S.; Schlemmer, K.H.; Straub, A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—An oral, direct Factor Xa inhibitor. J. Thromb. Haemost. 2005, 3, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Taune, V.; Skeppholm, M.; Agren, A.; Gryfelt, G.; Malmstrom, R.E.; Wikman, A.; Van Ryn, J.; Wallen, H. Rapid determination of anticoagulating effects of dabigatran in whole blood with rotational thromboelastometry and a thrombin-based trigger. J. Thromb. Haemost. 2018, 16, 2462–2470. [Google Scholar] [CrossRef]
- Wong, P.C.; Crain, E.J.; Xin, B.; Wexler, R.R.; Lam, P.Y.; Pinto, D.J.; Luettgen, J.M.; Knabb, R.M. Apixaban, an oral, direct and highly selective factor Xa inhibitor: In vitro, antithrombotic and antihemostatic studies. J. Thromb. Haemost. 2008, 6, 820–829. [Google Scholar] [CrossRef]
- Hemker, H.C.; Al Dieri, R.; De Smedt, E.; Beguin, S. Thrombin generation, a function test of the haemostatic-thrombotic system. Thromb. Haemost. 2006, 96, 553–561. [Google Scholar]
- Born, G.V.R. Aggregation of Blood Platelets by Adenosine Diphosphate and its Reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Soslau, G.; Class, R.; Morgan, D.A.; Foster, C.; Lord, S.T.; Marchese, P.; Ruggeri, Z.M. Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib. J. Biol. Chem. 2001, 276, 21173–21183. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef] [PubMed]
- Fadraersada, J.; Alva-Gallegos, R.; Skorepa, P.; Musil, F.; Javorska, L.; Matousova, K.; Krcmova, L.K.; Paclikova, M.; Carazo, A.; Blaha, V.; et al. Head-to-head ex vivo comparison of clinically used direct anticoagulant drugs. Naunyn Schmiedebergs Arch. Pharmacol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Eikelboom, J.; Oldgren, J.; Parekh, A.; Pogue, J.; Reilly, P.A.; Themeles, E.; Varrone, J.; et al. Dabigatran versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 361, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Hohnloser, S.H.; Oldgren, J.; Yang, S.; Wallentin, L.; Ezekowitz, M.; Reilly, P.; Eikelboom, J.; Brueckmann, M.; Yusuf, S.; Connolly, S.J. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Circulation 2012, 125, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Uchino, K.; Hernandez, A.V. Dabigatran association with higher risk of acute coronary events: Meta-analysis of noninferiority randomized controlled trials. Arch. Intern. Med. 2012, 172, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Petzold, T.; Thienel, M.; Konrad, I.; Schubert, I.; Regenauer, R.; Hoppe, B.; Lorenz, M.; Eckart, A.; Chandraratne, S.; Lennerz, C.; et al. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci. Transl. Med. 2016, 8, 367ra168. [Google Scholar] [CrossRef] [PubMed]
- Aleil, B.; Meyer, N.; Wolff, V.; Kientz, D.; Wiesel, M.L.; Gachet, C.; Cazenave, J.P.; Lanza, F. Plasma glycoprotein V levels in the general population: Normal distribution, associated parameters and implications for clinical studies. Thromb. Haemost. 2006, 96, 505–511. [Google Scholar]
- Giugliano, R.P.; Ruff, C.T.; Braunwald, E.; Murphy, S.A.; Wiviott, S.D.; Halperin, J.L.; Waldo, A.L.; Ezekowitz, M.D.; Weitz, J.I.; Spinar, J.; et al. Edoxaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2013, 369, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef]
- Patel, M.R.; Mahaffey, K.W.; Garg, J.; Pan, G.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; Hankey, G.J.; Piccini, J.P.; et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 2011, 365, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Pastori, D.; Bartimoccia, S.; Menichelli, D.; Vicario, T.; Nocella, C.; Carnevale, R.; Violi, F. Anti Xa oral anticoagulants inhibit in vivo platelet activation by modulating glycoprotein VI shedding. Pharmacol. Res. 2016, 113, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Dirienzo, L.; Vitulli, A.; Mancazzo, F.; Ammollo, C.T.; Dellanoce, C.; Paoletti, O.; Testa, S.; Colucci, M. Differential effect of direct oral anticoagulants on thrombin generation and fibrinolysis in patients with atrial fibrillation and venous thromboembolism. Blood Transfus. 2022, 20, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.R.; Castellucci, L.A.; Siegal, D.; Carrier, M. DOAC-associated bleeding, hemostatic strategies, and thrombin generation assays–a review of the literature. J. Thromb. Haemost. 2023, 21, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Arantes, F.B.B.; Menezes, F.R.; Franci, A.; Barbosa, C.; Dalcoquio, T.F.; Nakashima, C.A.K.; Baracioli, L.M.; Furtado, R.H.M.; Nomelini, Q.S.S.; Ramires, J.A.F.; et al. Influence of Direct Thrombin Inhibitor and Low Molecular Weight Heparin on Platelet Function in Patients with Coronary Artery Disease: A Prospective Interventional Trial. Adv. Ther. 2020, 37, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Ezekowitz, M.D.; Reilly, P.A.; Nehmiz, G.; Simmers, T.A.; Nagarakanti, R.; Parcham-Azad, K.; Pedersen, K.E.; Lionetti, D.A.; Stangier, J.; Wallentin, L. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO Study). Am. J. Cardiol. 2007, 100, 1419–1426. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dabigatran | Rivaroxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| ETP (nM × min) | 1918 (1202–2591) | 965 (600–2048) § | 808 (539 –1516) # | 642 (406–1118) * | 1919 (1203–2591) | 1047 (773–1725) § | 806 (647–1408) ° | 765 (555–1342) * |
| Peak (nM) | 240 (147–285) | 133 (100–298) | 200 (130–271) | 181 (68–216) | 227 (128–278) | 67 (49–88) * | 52 (36–88) * | 49 (32–76) * |
| Time to peak (min) | 6.2 (5.6–7.7) | 4.7 (3.7–7.0) | 4.7 (3.3–11.4) | 4.0 (3.2–12.9) | 6.2 (5.6–7.7) | 13.0 (7.8–17.2) ° | 12.5 (9.8–20.9) * | 15.9 (13.2–18.6) * |
| Lag time (min) | 1.33 (0.7–2.4) | 2.3 (1.5–3.2) | 2.3 (0.7–6.9) | 2.0 (1.0–6.8) | 1.3 (0.7–2.4) | 3.0 (1.6–4.8) § | 4.5 (2.9–6.5) * | 5.8 (4.4–7.6) * |
| Apixaban | Edoxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| ETP (nM × min) | 1918 (1202–2591) | 1158 (780–1934) § | 1100 (697–1482) # | 949 (520–1295) ° | 1218 (975–1794) | 1118 (853–1720) | 938 (805–1464) | 819 (734–1039) § |
| Peak (nM) | 227 (128–278) | 77 (50–136) * | 62 (40–88) * | 49 (38–70) * | 101 (85–142) | 97 (63–182) | 62 (53–96) § | 50 (39–69) ° |
| Time to peak (min) | 6.2 (5.6–7.7) | 10 (6.6–14.3) § | 14 (9.8–20.6) * | 15 (12–19) * | 11 (9–13) | 14 (12–17) § | 18 (15–20) * | 19 (17–22) * |
| Lag time (min) | 1.3 (0.7–2.4) | 2.7 (1.7–4.5) § | 3.3 (1.6–6.0) # | 4.3 (3.6–6.8) * | 5.3 (4.9–7.8) | 8.2 (6.0–10.2) § | 11 (7.4–13.3) ° | 12 (10.0–13.9) * |
| LTA (% of Maximum) | Dabigatran | Rivaroxaban | ||||||
| Inducing Agent | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| ADP | 73 (50−77) | 72 (30−76) | 54 (39−80) | 68 (24−83) | 73 (38−82) | 50 (31−82) | 75 (35−82) | 69 (34−84) |
| TRAP | 70 (66−79) | 76 (72−78) | 71 (59−73) | 72 (60−76) | 80 (44−85) | 49 (32−88) | 36 (26−81) | 80 (32−89) |
| Thrombin | 82 (64−93) | 0 (0−0) * | 0 (0−0) * | 0 (0−0) * | 75 (65−90) | 78 (72−85) | 80 (75−88) | 78 (70−84) |
| Tissue Factor | NA | NA | NA | NA | 85 (80−91) | 0 (0−60) * | 0 (0−60) * | 0 (0−70) * |
| LTA (% of maximum) | Apixaban | Edoxaban | ||||||
| Inducing Agent | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| ADP | 73 (38−82) | 73 (39−82) | 80 (30−85) | 78 (40−86) | 76 (82−90) | 82 (76−89) | 86 (73−94) | 79 (70−91) |
| TRAP | 80 (44−85) | 80 (34−88) | 55 (23−82) | 72 (34−81) | 85 (80−98) | 83 (80−93) | 81 (73−93) | 85 (78−96) |
| Thrombin | 75 (65−90) | 76 (73−82) | 78 (70−82) | 80 (76−84) | 91 (77−97) | 91 (80−96) | 82 (70−96) | 88 (78−97) |
| Tissue Factor | 85 (80−91) | 18 (0−59) * | 10 (0−56) * | 0 (0−31) * | 81 (78−94) | 66 (50−83) | 53 (25−90) # | 36 (7−56) * |
| Dabigatran | Rivaroxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| Serum TXB2 (ng/mL) | 649 (500−708) | 439 (258−1098) | 457 (253−1070) | 158 (129−470) # | 581 (431−860) | 467 (130−568) | 373 (124−507) § | 137 (88−338) # |
| Apixaban | Edoxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| Serum TXB2 (ng/mL) | 581 (431−860) | 514 (341−586) | 332 (262−608) | 354 (246−548) § | 507 (318−627) | 428 (319−624) | 402 (278−555) | 400 (239−488) # |
| Dabigatran | Rivaroxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| PAR-1 (MFI ratio) | 22.1 (21.4−31.1) | 74.2 (44.5−118.9) | 127.8 (46.9−159.8) § | 127.9 (48.4−186.5) ° | 5.7 (4.6−7.0) | 5.7 (4.2−7.4) | 5.7 (5.0−8.0) | 6.0 (3.9−7.3) |
| CD62+/CD41+ (MFI %) | 0.6 (0.1−4.8) | 0.8 (0.3−3.9) | 0.6 (3.4−1.9) | 0.5 (0.1−2.8) | 22 (10−27) | 16 (10−21) | 15 (9−24) | 17 (10−31) |
| Apixaban | Edoxaban | |||||||
| Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | Control | 50 ng/mL | 150 ng/mL | 250 ng/mL | |
| median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | median (IQR) | |
| PAR-1 (MFI ratio) | 5.7 (4.6−7.0) | 5.7 (4.5−7.9) | 5.7 (4.3−6.8) | 5.4 (4.3−6.7) | 3.1 (2.0−4.7) | 3.6 (2.0−4.8) | 3.9 (2.1−4.8) | 3.6 (2.1−4.6) |
| CD62+/CD41+ (MFI %) | 22 (10−27) | 19 (10−32) | 18 (11−26) | 16 (13−27) | 27 (19−48) | 30 (20−49) | 32 (22−49) | 33 (25−49) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renda, G.; Bucciarelli, V.; Barbieri, G.; Lanuti, P.; Berteotti, M.; Malatesta, G.; Cesari, F.; Salvatore, T.; Giusti, B.; Gori, A.M.; et al. Ex Vivo Antiplatelet Effects of Oral Anticoagulants. J. Cardiovasc. Dev. Dis. 2024, 11, 111. https://doi.org/10.3390/jcdd11040111
Renda G, Bucciarelli V, Barbieri G, Lanuti P, Berteotti M, Malatesta G, Cesari F, Salvatore T, Giusti B, Gori AM, et al. Ex Vivo Antiplatelet Effects of Oral Anticoagulants. Journal of Cardiovascular Development and Disease. 2024; 11(4):111. https://doi.org/10.3390/jcdd11040111
Chicago/Turabian StyleRenda, Giulia, Valentina Bucciarelli, Giulia Barbieri, Paola Lanuti, Martina Berteotti, Gelsomina Malatesta, Francesca Cesari, Tanya Salvatore, Betti Giusti, Anna Maria Gori, and et al. 2024. "Ex Vivo Antiplatelet Effects of Oral Anticoagulants" Journal of Cardiovascular Development and Disease 11, no. 4: 111. https://doi.org/10.3390/jcdd11040111
APA StyleRenda, G., Bucciarelli, V., Barbieri, G., Lanuti, P., Berteotti, M., Malatesta, G., Cesari, F., Salvatore, T., Giusti, B., Gori, A. M., Marcucci, R., & De Caterina, R. (2024). Ex Vivo Antiplatelet Effects of Oral Anticoagulants. Journal of Cardiovascular Development and Disease, 11(4), 111. https://doi.org/10.3390/jcdd11040111