
Haplotype-Phased Chromosome-Level Genome Assembly of Cryptoporus qinlingensis, a Typical Traditional Chinese Medicine Fungus



Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Material and Nucleic Acid Extraction
2.2. Genome Sequencing, De Novo Assembly, Annotation, and Visualization
2.2.1. NGS, PacBio, and RNA-Seq Library Construction and Sequencing
2.2.2. De Novo Assembly, Haplotype Phasing, and Hi-C Scaffolding
2.2.3. Gene Prediction and Genome Annotation
2.2.4. Genomic Circular Map
2.2.5. Comparative Genomics Analysis
2.2.6. Repeat Sequence Identification
2.3. Phylogenomic Analysis and Gene Family Variation Analysis
2.4. CAZy Family and Cytochrome P450 Analyses
2.5. BGC Analysis and Visualization
2.6. Data Availability
3. Results
3.1. Chromosome-Level Genome Assembly and Haplotype-Phasing of Cryptoporus
3.2. Genome Annotation and Comparative Genome Analysis
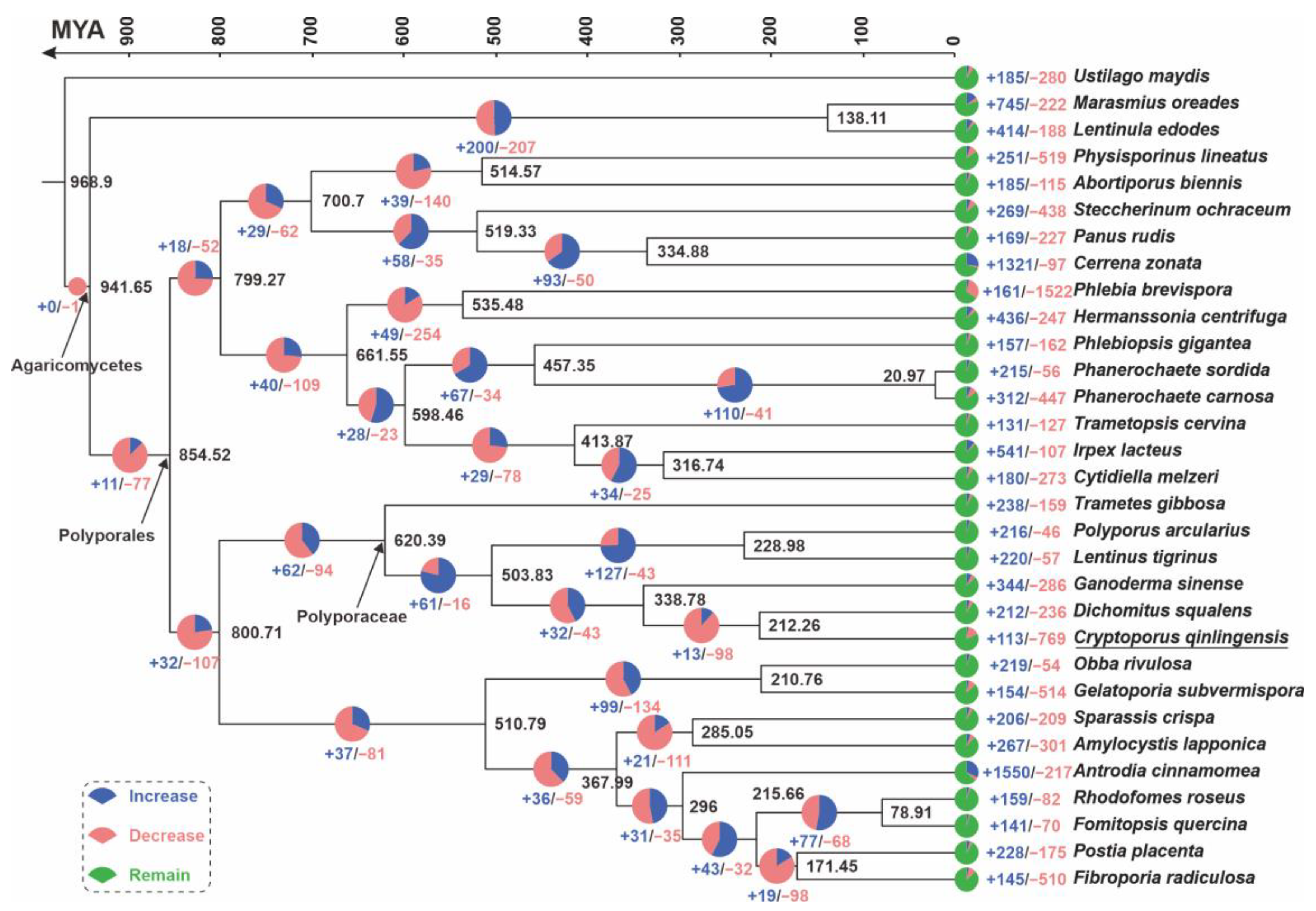
3.3. Phylogenetic and Gene Family Variation Analysis
3.4. CAZyme Analysis
3.5. Identifying Mating Genes and Developing SSR Markers
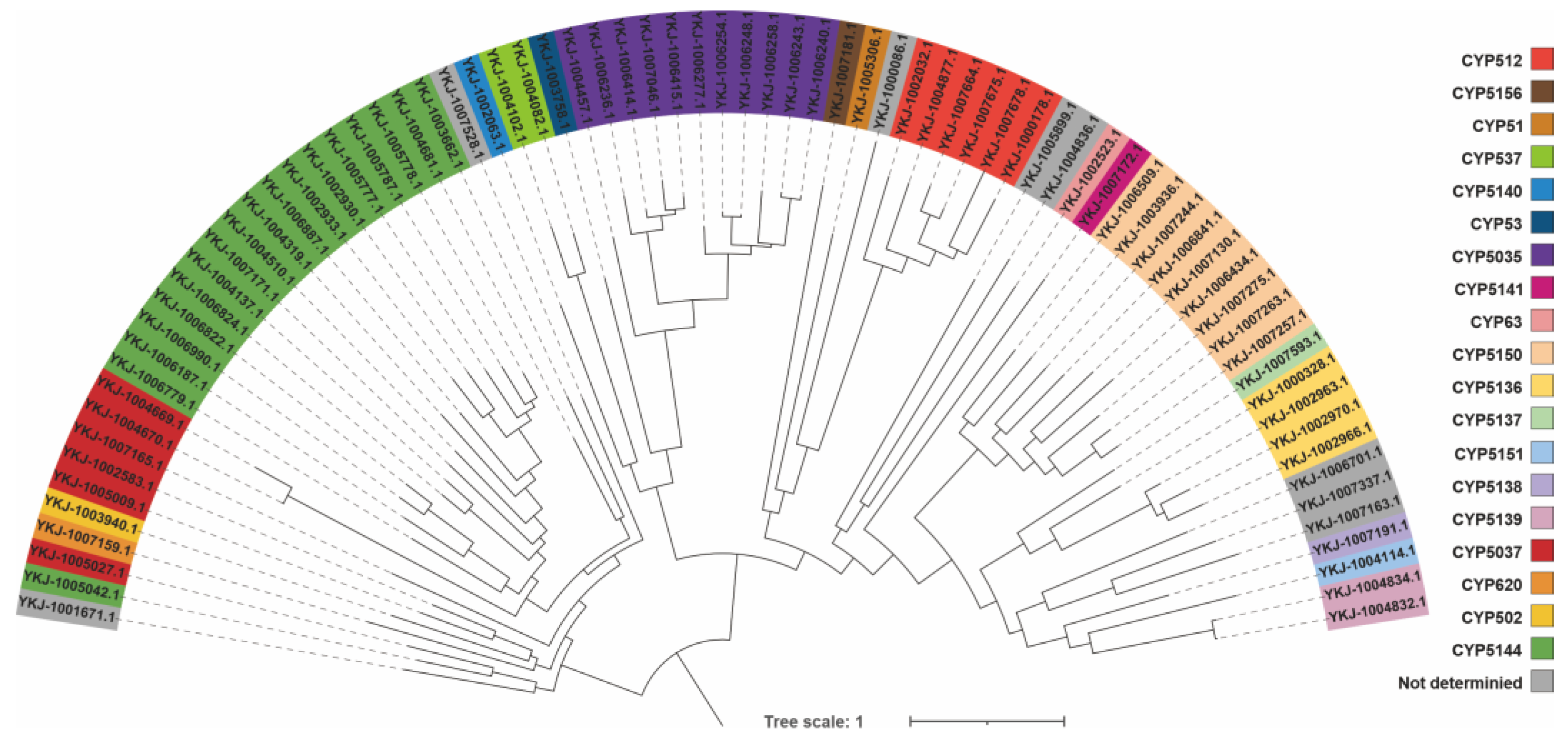
3.6. Analysis and Identification of Transcription Factors and P450 Genes in C. qinlingensis
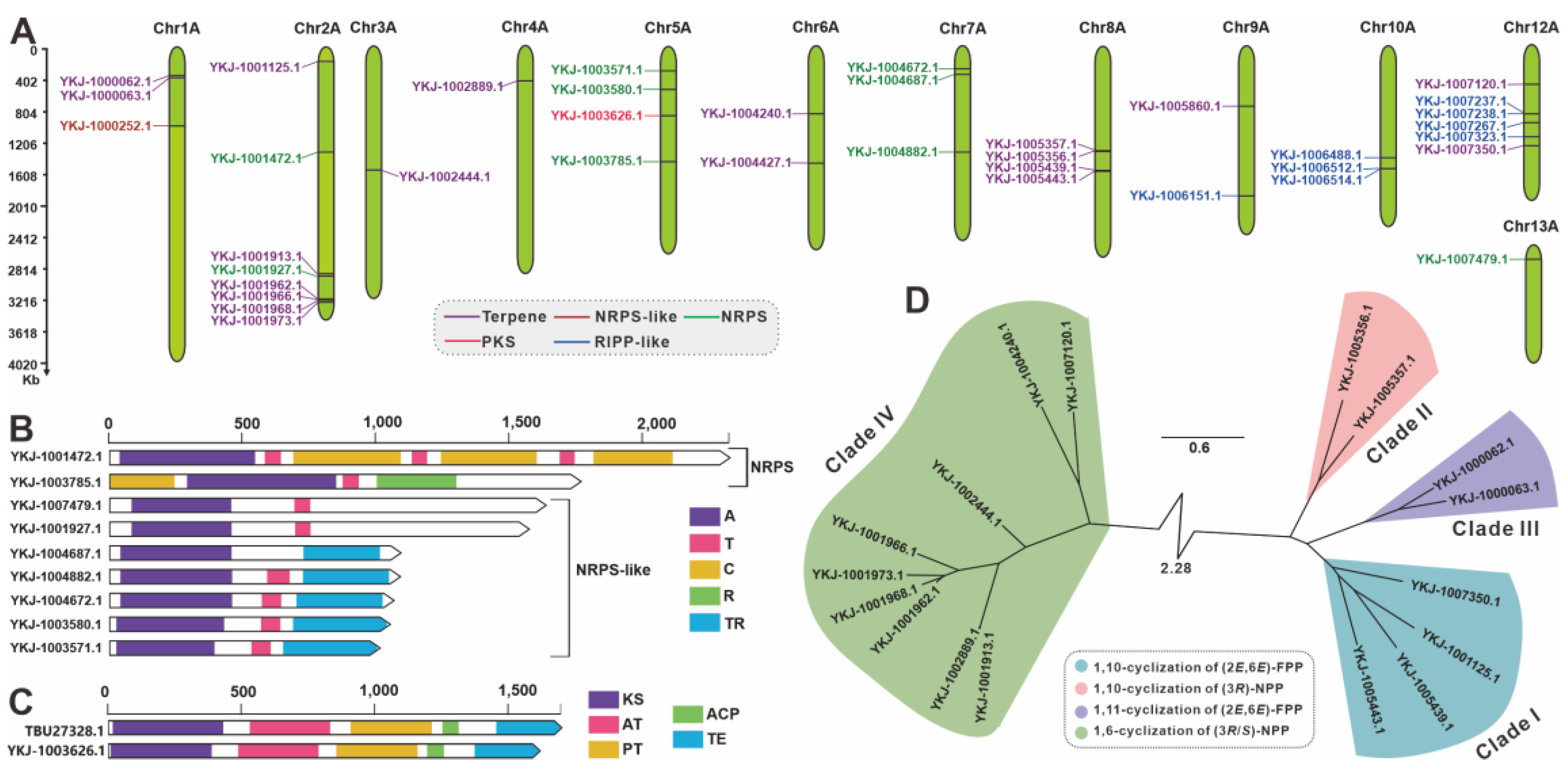
3.7. Search and Analysis of Genes (Clusters) Involved in Secondary Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, L.Y.; Yu, X.H.; Lu, B.; Hua, Y. Bioassay-Guided Isolation of Cytotoxic Isocryptoporic Acids from Cryptoporus volvatus. Molecules 2016, 21, 1692. [Google Scholar] [CrossRef] [PubMed]
- Jiandong, Z. Flora fungorum sinicorum In Polyporaceae; Science Press: Beijing, China, 1998; Volume 3, pp. 88–89. [Google Scholar]
- Song, Y.; Wang, Y.-Y.; Wang, Y.; Xie, X.-C. Cryptoporus qinlingensis sp. nov., the third species of Cryptoporus (Polyporaceae, Polyporales) from northwestern China. Phytotaxa 2024, 672, 242–254. [Google Scholar] [CrossRef]
- Hirotani, M.; Furuya, T.; Shiro, M. Cryptoporic acids H and I, drimane sesquiterpenes from Ganoderma neo-japonicum and Cryptoporus volvatus. Phytochemistry 1991, 30, 1555–1559. [Google Scholar] [CrossRef]
- Wu, W.; Zhao, F.; Ding, R.; Bao, L.; Gao, H.; Lu, J.-C.; Yao, X.-S.; Zhang, X.-Q.; Liu, H.-W. Four New Cryptoporic Acid Derivatives from the Fruiting Bodies of Cryptoporus sinensis, and Their Inhibitory Effects on Nitric Oxide Production. Chem. Biodivers. 2011, 8, 1529–1538. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Gao, L.; Cao, L.; Lv, N.; Shen, L.; Si, J. Two new cryptoporic acid derivatives from the fruiting bodies of Cryptoporus volvatus. Phytochem. Lett. 2015, 14, 63–66. [Google Scholar] [CrossRef]
- Wang, J.C.; Li, G.Z.; Lv, N.; Shen, L.G.; Shi, L.L.; Si, J.Y. Cryptoporic acid S, a new drimane-type sesquiterpene ether of isocitric acid from the fruiting bodies of Cryptoporus volvatus. J. Asian Nat. Prod. Res. 2017, 19, 719–724. [Google Scholar] [CrossRef]
- Duan, C.; Ge, X.; Wang, J.; Wei, Z.; Feng, W.H.; Wang, J. Ergosterol peroxide exhibits antiviral and immunomodulatory abilities against porcine deltacoronavirus (PDCoV) via suppression of NF-κB and p38/MAPK signaling pathways in vitro. Int. Immunopharmacol. 2021, 93, 107317. [Google Scholar] [CrossRef]
- Duan, C.; Wang, J.; Liu, Y.; Zhang, J.; Si, J.; Hao, Z.; Wang, J. Antiviral effects of ergosterol peroxide in a pig model of porcine deltacoronavirus (PDCoV) infection involves modulation of apoptosis and tight junction in the small intestine. Vet. Res. 2021, 52, 86. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Wang, J.; Zhang, J.; Duan, C.; Wang, J. Ergosterol Peroxide Inhibits Porcine Epidemic Diarrhea Virus Infection in Vero Cells by Suppressing ROS Generation and p53 Activation. Viruses 2022, 14, 402. [Google Scholar] [CrossRef]
- Xie, Q.M.; Deng, J.F.; Deng, Y.M.; Shao, C.S.; Zhang, H.; Ke, C.K. Effects of cryptoporus polysaccharide on rat allergic rhinitis associated with inhibiting eotaxin mRNA expression. J. Ethnopharmacol. 2006, 107, 424–430. [Google Scholar] [CrossRef]
- Sodeinde, K.O.; Adeoye, A.O.; Adesipo, A.; Adeniyi, A.A.; Falode, J.A.; Obafemi, T.O.; Olusanya, S.O.; Twigge, L.; Conradie, J.; Mosaku, T.O. Isolation, characterization and modulatory potentials of β-stigmasterol, ergosterol and xylopic acid from Anchomanes difformis on mitochondrial permeability transition pore in vitro. Chin. Herb. Med. 2023, 15, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.F.; Chen, J.Q.; Xie, Q.M.; Zhao, X.Y.; Ke, C.K. Effects of polysaccharides of Cryptoporus volvatus on bronchial hyperreasponsiveness and inflammatory cells in ovalbumin sensitized rats. Zhejiang Da Xue Xue Bao Yi Xue Ban 2003, 32, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.Y.; Zhang, L.H.; Shen, J.; Shen, H.J.; Jia, Y.L.; Yan, X.F.; Xie, Q.M. Cyptoporus polysaccharide prevents lipopolysaccharide-induced acute lung injury associated with down-regulating Toll-like receptor 2 expression. J. Ethnopharmacol. 2011, 137, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, W.; Guo, J.; Huang, N. Composition Analysis of Volatile Oil in Wild Cryptoporus volvatus. J. Fujian Coll. TCM 1998, 8, 26–28. [Google Scholar]
- Ma, Z.; Zhang, W.; Wang, L.; Zhu, M.; Wang, H.; Feng, W.H.; Ng, T.B. A novel compound from the mushroom Cryptoporus volvatus inhibits porcine reproductive and respiratory syndrome virus (PRRSV) in vitro. PLoS ONE 2013, 8, e79333. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, W.; Sun, Y.; Yang, Q.; Ren, J.; Liu, J.; Wang, H.; Feng, W.H. Cryptoporus volvatus extract inhibits porcine reproductive and respiratory syndrome virus (PRRSV) in vitro and in vivo. PLoS ONE 2013, 8, e63767. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, Z.; Xiong, F.; Chen, Y.; Sun, Y. Anti-tumor mechanism of sesquiterpenoids from Cryptoporus volvatus based on molecular docking. Nan Fang Yi Ke Da Xue Xue Bao 2022, 42, 71–77. [Google Scholar] [CrossRef]
- Henn, M.R.; Gleixner, G.; Chapela, I.H. Growth-dependent stable carbon isotope fractionation by basidiomycete fungi: δ13C pattern and physiological process. Appl. Env. Microbiol. 2002, 68, 4956–4964. [Google Scholar] [CrossRef]
- Zhang, R.-Q.; Feng, X.-L.; Wang, Z.-X.; Xie, T.-C.; Duan, Y.; Liu, C.; Gao, J.-M.; Qi, J. Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application. J. Fungi 2022, 8, 1245. [Google Scholar] [CrossRef]
- Leiter, É.; Emri, T.; Pákozdi, K.; Hornok, L.; Pócsi, I. The impact of bZIP Atf1ortholog global regulators in fungi. Appl. Microbiol. Biotechnol. 2021, 105, 5769–5783. [Google Scholar] [CrossRef]
- Durairaj, P.; Hur, J.-S.; Yun, H. Versatile biocatalysis of fungal cytochrome P450 monooxygenases. Microb. Cell Factories 2016, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Hatakka, A.; Hammel, K.E. Fungal Biodegradation of Lignocelluloses. In Industrial Applications; Hofrichter, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 319–340. [Google Scholar]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Kou, Z.-x.; Yao, Y.-h.; Hu, Y.-f.; Zhang, B.-p. Discussion on position of China’s north-south transitional zone by comparative analysis of mountain altitudinal belts. J. Mt. Sci. 2020, 17, 1901–1915. [Google Scholar] [CrossRef]
- Xie, X.; Zhao, L.; Song, Y.; Qiao, Y.; Wang, Z.-X.; Qi, J. Genome-wide characterization and metabolite profiling of Cyathus olla: Insights into the biosynthesis of medicinal compounds. BMC Genom. 2024, 25, 618. [Google Scholar] [CrossRef]
- He, X.; Huo, W.; Zhang, L.; Dai, L.; Liu, Y.; Li, J. Two New Species of Helvella (Pezizales, Ascomycota) Collected from the Qinling Mountains of China. J. Fungal Res. 2024, 22, 226–235. [Google Scholar] [CrossRef]
- He, X.; Huo, W.; Zhang, L.; Dai, L.; Qi, P.; Liu, Y.; Li, J. A New Species of Hygrophoropsis (Boletales, Basidiomycota) from Qinling Mountains in China. J. Fungal Res. 2024, 22, 135–141. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Huang, K.; Pu, K.-L.; Li, L.; Jiang, W.-X.; Wu, J.; Kawagishi, H.; Li, M.; Qi, J. Naematelia aurantialba: A comprehensive review of its medicinal, nutritional, and cultivation aspects. Food Med. Homol. 2025. [Google Scholar] [CrossRef]
- Abdel-Wareth, M.T.A. Fungal Secondary Metabolites: Current Research, Commercial Aspects, and Applications. In Industrially Important Fungi for Sustainable Development: Volume 2: Bioprospecting for Biomolecules; Abdel-Azeem, A.M., Yadav, A.N., Yadav, N., Sharma, M., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 299–346. [Google Scholar]
- Vivek-Ananth, R.P.; Sahoo, A.K.; Kumaravel, K.; Mohanraj, K.; Samal, A. MeFSAT: A curated natural product database specific to secondary metabolites of medicinal fungi. RSC Adv. 2021, 11, 2596–2607. [Google Scholar] [CrossRef]
- Qi, J.; Gao, Y.-Q.; Kang, S.-j.; Liu, C.; Gao, J.-M. Secondary metabolites of bird’s nest fungi: Chemical structures and biological activities. J. Agric. Food Chem. 2023, 71, 6513–6524. [Google Scholar] [CrossRef]
- Qi, J.; Wu, J.; Kang, S.; Gao, J.; Hirokazu, K.; Liu, H.; Liu, C. The chemical structures, biosynthesis, and biological activities of secondary metabolites from the culinary-medicinal mushrooms of the genus Hericium: A review. Chin. J. Nat. Med. 2024, 22, 676–698. [Google Scholar] [CrossRef]
- Lee, I.-K.; Yun, B.-S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Zhen-xin, W.; Xi-long, F.; Chengwei, L.; Gao, J.-m.; Jianzhao, Q. Diverse Metabolites and Pharmacological Effects from the Basidiomycetes Inonotus hispidus. Antibiotics 2022, 11, 1097. [Google Scholar] [CrossRef]
- Takahashi, H.; Toyota, M.; Asakawa, Y. Drimane-type sesquiterpenoids from Cryptoporus volvatus infected by Paecilomyces varioti. Phytochemistry 1993, 33, 1055–1059. [Google Scholar] [CrossRef]
- Asakawa, Y.; Hashimoto, T.; Mizuno, Y.; Tori, M.; Fukazawa, Y. Cryptoporic acids A-G, drimane-type sesquiterpenoid ethers of isocitric acid from the fungus Cryptoporus volvatus. Phytochemistry 1992, 31, 579–592. [Google Scholar] [CrossRef]
- Lackner, G.; Bohnert, M.; Wick, J.; Hoffmeister, D. Assembly of Melleolide Antibiotics Involves a Polyketide Synthase with Cross-Coupling Activity. Chem. Biol. 2013, 20, 1101–1106. [Google Scholar] [CrossRef]
- Braesel, J.; Fricke, J.; Schwenk, D.; Hoffmeister, D. Biochemical and genetic basis of orsellinic acid biosynthesis and prenylation in a stereaceous basidiomycete. Fungal Genet. Biol. 2017, 98, 12–19. [Google Scholar] [CrossRef]
- Yu, P.W.; Chang, Y.C.; Liou, R.F.; Lee, T.H.; Tzean, S.S. pks63787, a polyketide synthase gene responsible for the biosynthesis of benzenoids in the medicinal mushroom Antrodia cinnamomea. Planta Medica 2016, 82, 2. [Google Scholar]
- Han, H.; Yu, C.; Qi, J.; Wang, P.; Zhao, P.; Gong, W.; Xie, C.; Xia, X.; Liu, C. High-efficient production of mushroom polyketide compounds in a platform host Aspergillus oryzae. Microb. Cell Factories 2023, 22, 60. [Google Scholar] [CrossRef]
- Harvey, C.J.B.; Tang, M.; Schlecht, U.; Horecka, J.; Fischer, C.R.; Lin, H.C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M.; et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, 14. [Google Scholar] [CrossRef]
- Ishiuchi, K.; Nakazawa, T.; Ookuma, T.; Sugimoto, S.; Sato, M.; Tsunematsu, Y.; Ishikawa, N.; Noguchi, H.; Hotta, K.; Moriya, H.; et al. Establishing a New Methodology for Genome Mining and Biosynthesis of Polyketides and Peptides through Yeast Molecular Genetics. Chembiochem 2012, 13, 846–854. [Google Scholar] [CrossRef]
- Löhr, N.A.; Eisen, F.; Thiele, W.; Platz, L.; Motter, J.; Hüttel, W.; Gressler, M.; Müller, M.; Hoffmeister, D. Unprecedented Mushroom Polyketide Synthases Produce the Universal Anthraquinone Precursor. Angew. Chem.-Int. Ed. 2022, 61, 6. [Google Scholar] [CrossRef] [PubMed]
- Löhr, N.A.; Urban, M.C.; Eisen, F.; Platz, L.; Hüttel, W.; Gressler, M.; Müller, M.; Hoffmeister, D. The Ketosynthase Domain Controls Chain Length in Mushroom Oligocyclic Polyketide Synthases. Chembiochem 2023, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Löhr, N.A.; Rakhmanov, M.; Wurlitzer, J.M.; Lackner, G.; Gressler, M.; Hoffmeister, D. Basidiomycete non-reducing polyketide synthases function independently of SAT domains. Fungal Biol. Biotechnol. 2023, 10, 17. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster No. | Location | Start (bp) | End (bp) | Core Gene ID | Core Gene Type |
|---|---|---|---|---|---|
| 1 | Chr1A | 359,344 | 398,787 | YKJ-1000062.1 | Terpene |
| 2 | Chr1A | 999,437 | 1,046,045 | YKJ-1000252.1 | β-lactone |
| YKJ-1000255.1 | NRPS-like | ||||
| 3 | Chr2A | 174,241 | 195,721 | YKJ-1001125.1 | Terpene |
| 4 | Chr2A | 2,883,761 | 2,905,036 | YKJ-1001913.1 | Terpene |
| 5 | Chr2A | 3,211,700 | 3,266,305 | YKJ-1001962.1 | Terpene |
| 6 | Chr2A | 1,315,680 | 1,363,525 | YKJ-1001472.1 | NRPS |
| 7 | Chr2A | 2,907,662 | 2,952,731 | YKJ-1001927.1 | NRPS-like |
| 8 | Chr3A | 1,573,377 | 1,594,751 | YKJ-1002444.1 | Terpene |
| 9 | Chr4A | 443,477 | 464,752 | YKJ-1002889.1 | Terpene |
| 10 | Chr5A | 288,054 | 332,091 | YKJ-1003571.1 | NRPS-like |
| 11 | Chr5A | 530,357 | 574,388 | YKJ-1003580.1 | NRPS-like |
| 12 | Chr5A | 1,444,291 | 1,491,423 | YKJ-1003785.1 | NRPS |
| 13 | Chr5A | 862,970 | 908,215 | YKJ-1003626.1 | PKS |
| 14 | Chr6A | 855,429 | 876,688 | YKJ-1004240.1 | Terpene |
| 15 | Chr6A | 1,486,316 | 1,507,452 | YKJ-1004427.1 | Terpene |
| 16 | Chr7A | 272,998 | 317,249 | YKJ-1004672.1 | NRPS-like |
| 17 | Chr7A | 340,327 | 384,582 | YKJ-1004687.1 | NRPS-like |
| 18 | Chr7A | 1,337,635 | 1,381,910 | YKJ-1004882.1 | NRPS-like |
| 19 | Chr8A | 1,324,708 | 1,349,324 | YKJ-1005356.1 | Terpene |
| YKJ-1005357.1 | Terpene | ||||
| 20 | Chr8A | 1,567,264 | 1,600,522 | YKJ-1005439.1 | Terpene |
| YKJ-1005443.1 | Terpene | ||||
| 21 | Chr9A | 761,639 | 783,524 | YKJ-1005860.1 | Terpene |
| 22 | Chr9A | 1,894,286 | 1,954,964 | YKJ-1006151.1 | RiPP |
| 23 | Chr10A | 1,393,108 | 1,453,799 | YKJ-1006488.1 | RiPP |
| 24 | Chr10A | 1,533,952 | 1,597,764 | YKJ-1006512.1 | RiPP |
| YKJ-1006514.1 | RiPP | ||||
| 25 | Chr12A | 498,674 | 520,051 | YKJ-1007120.1 | Terpene |
| 26 | Chr12A | 1,271,344 | 1,292,712 | YKJ-1007350.1 | Terpene |
| 27 | Chr12A | 849,229 | 911,262 | YKJ-1007237.1 | RiPP |
| YKJ-1007238.1 | RiPP | ||||
| 28 | Chr12A | 957,737 | 1,018,469 | YKJ-1007267.1 | RiPP |
| 29 | Chr12A | 1,147,393 | 1,208,064 | YKJ-1007323.1 | RiPP |
| 30 | Chr13A | 155,448 | 200,573 | YKJ-1007479.1 | NRPS-like |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Zhang, M.; Liu, Y.-Y.; Li, M.; Xie, X.; Qi, J. Haplotype-Phased Chromosome-Level Genome Assembly of Cryptoporus qinlingensis, a Typical Traditional Chinese Medicine Fungus. J. Fungi 2025, 11, 163. https://doi.org/10.3390/jof11020163
Song Y, Zhang M, Liu Y-Y, Li M, Xie X, Qi J. Haplotype-Phased Chromosome-Level Genome Assembly of Cryptoporus qinlingensis, a Typical Traditional Chinese Medicine Fungus. Journal of Fungi. 2025; 11(2):163. https://doi.org/10.3390/jof11020163
Chicago/Turabian StyleSong, Yu, Ming Zhang, Yu-Ying Liu, Minglei Li, Xiuchao Xie, and Jianzhao Qi. 2025. "Haplotype-Phased Chromosome-Level Genome Assembly of Cryptoporus qinlingensis, a Typical Traditional Chinese Medicine Fungus" Journal of Fungi 11, no. 2: 163. https://doi.org/10.3390/jof11020163
APA StyleSong, Y., Zhang, M., Liu, Y.-Y., Li, M., Xie, X., & Qi, J. (2025). Haplotype-Phased Chromosome-Level Genome Assembly of Cryptoporus qinlingensis, a Typical Traditional Chinese Medicine Fungus. Journal of Fungi, 11(2), 163. https://doi.org/10.3390/jof11020163