Abstract
Armillaria species have a global distribution and play various roles in the natural ecosystems, e.g., pathogens, decomposers, and mycorrhizal associates. However, their taxonomic boundaries, speciation processes, and origin are poorly understood. Here, we used a phylogenetic approach with 358 samplings from Europe, East Asia, and North America to delimit the species boundaries and to discern the evolutionary forces underpinning divergence and evolution. Three species delimitation methods indicated multiple unrecognized phylogenetic species, and biological species recognition did not reflect the natural evolutionary relationships within Armillaria; for instance, biological species of A. mellea and D. tabescens are divergent and cryptic species/lineages exist associated with their geographic distributions in Europe, North America, and East Asia. While the species-rich and divergent Gallica superclade might represent three phylogenetic species (PS I, PS II, and A. nabsnona) that undergo speciation. The PS II contained four lineages with cryptic diversity associated with the geographic distribution. The genus Armillaria likely originated from East Asia around 21.8 Mya in early Miocene when Boreotropical flora (56–33.9 Mya) and the Bering land bridge might have facilitated transcontinental dispersal of Armillaria species. The Gallica superclade arose at 9.1 Mya and the concurrent vicariance events of Bering Strait opening and the uplift of the northern Tibetan plateau might be important factors in driving the lineage divergence.
1. Introduction
Understanding the biogeographical pattern and origin of fungi is important, especially for those fungal plant pathogens that cause severe economic losses. However, studying fungal biogeography is challenging. This has been attributed to shortcomings in delimiting species based on morphological characteristics, poor knowledge of the phylogeny in most fungal groups, rare fossil records, and the long-distance dispersal ability of spores to overcome geographic barriers [1]. For example, it took over a century of efforts to figure out the origin and dispersal pattern of Pyricularia oryzae, the famous pathogen of rice blast, since it was first discovered in 1892 [2,3]. Benefiting from the development of DNA technology and molecular analysis methods, there has been a breakthrough of fungal biogeography in recent decades [4,5,6]. Based on molecular calibration, the current distribution patterns and ancestral areas of many fungal groups have been well explained by vicariance events [7,8,9,10,11].
Armillaria species are well known as root rot pathogens, infecting over 500 host plants in both natural forests and silvicultural systems [12]. Individuals of Armillaria can reach immense sizes via growing rhizomorphs from one host plant to the neighboring plants, resulting in the largest known terrestrial organism in the world [13]. As mycorrhiza, some Armillaria species are associated with Gastrodia elata (“Tianma” in Chinese) [14] and Polyporus umbellatus that are widely used as traditional medicines in Asian countries [15]. The first biogeography study of Armillaria was reported in 2011, in which a non-Holarctic origin for Armillaria was suggested based on a phylogeny constructed by ITS, 28S, and TEF-1α [16]. In contrast, a recent study with a more robust multi-loci (28S, TEF-1α, rpb2, tub, gpd, and act) phylogeny of Armillaria inferred that Eurasia was the most probable origin [17]. Nevertheless, Eurasia is the combined continents of Europe and Asia, spanning from Asia Pacific to Atlantic Europe, and it is necessary to reconstruct the ancestral location of Armillaria.
The taxonomy of Armillaria has raised lots of controversial discussion. Currently, the accepted generic name is Armillaria Fr. Staude (1857) and A. mellea (Vahl: Fr.) P. Kumm is the type species [18,19]. Recently, Armillaria was narrowed down to include only annulate species, and Desarmillaria was introduced to accommodate two exannulate species (D. ectypa and D. tabescens) [17]. By now, a total of 278 Armillaria names are recorded in Index Fungorum (http://www.indexfungorum.org/Names/Names.asp) (accessed on 6 November 2021), but the validity of these name is often in debate [19]. On the delimitation of Armillaria species, the Biological Species Recognition (BSR) approach has prevailed since Korhonen [20] first employed this approach to delimit the biological species in the European population. Armillaria in many regions are recorded in biological species but they are not formally described, such as multiple Chinese Biological Species (CBS), CBS C, CBS F, CBS H, CBS J, CBS L, CBS N, CBS P, and CBS O [21]. Despite these limitations and weaknesses (e.g., ambiguous results and post-zygotic isolation not considered) in the practical application of BSR [12], these biological species represent great species diversity and are invaluable to understand the phylogeny of Armillaria. However, these biological species were not considered in previous studies of Armillaria phylogeography [16,17]. The Phylogenetic Species Concept (PSR) was introduced by Nixon and Wheeler, which emphasized the monophyly of species [22]. By using genealogical concordance phylogenetic species recognition (GCPSR), a large number of cryptic species in Fusarium [23,24] and Collectotrichum [25] have been uncovered under PSR. Similarly, phylogenetic analyses of DNA sequence data have been used to identify Armillaria strains to the species level, but the phylogenetic relationships of many biological species have not been clearly resolved [26,27].
The Armillaria genus has a worldwide distribution. Species occurring naturally in the northern hemisphere and southern hemisphere are phylogenetically distinct [28]. In the northern hemisphere, some species have transcontinental distributions, e.g., A. mellea, A. gallica, A. ostoyae, and D. tabescens (previously defined as A. tabescens), reported from Europe, Asia, and North America, and the North American D. tabescens has been separated as D. caespitosa recently [29]; A. borealis, A. cepistipes, and D. ectypa (previously defined as A. ectypa) distributed in both Europe and East Asia but not reported from North America; and A. nabsnona and A. sinapina distributed in both North America and East Asia [12,28,30]. Whereas, some other species are restricted to specific continents, such as A. gemina, A. altimontana, A. calvescens, and A. solidipes were only discovered in North America [31,32] and A. singula, A. jezoensis, and seven Chinese biological species were only known from East Asia [21,33]. Furthermore, there has been growing use of molecular approaches for species delimitation, such as the generalized mixed Yule-coalescent (GMYC) model [34,35], Poisson tree processes (PTP) model [36], and Bayesian multispecies coalescent method (BPP) [37]. These methods have not yet been applied in the phylogeny of Armillaria. Studying the species delimitation and relationships in Armillaria in the northern hemisphere would be useful to understand the genetic variability, as well as inter- and intraspecific divergence. The molecular data could infer the ecological factors and evolutionary processes that shape the various distribution patterns.
In this study, we collected 358 Armillaria samples in the northern hemisphere with an extensive distribution range covering Europe, North America, and Asia. First, we investigate the species boundaries and relationship of Armillaria spp. in the northern hemisphere according to GCPSR, PTP and BPP methods. Second, we aimed to answer the questions on the speciation event and evolutionary processes in Armillaria, such as allopatry or lineage divergence, due to geological and vicariance events and whether strains from different geographic regions could represent distinct species and have evolved independently. Third, we performed molecular dating to reconstruct the ancestral area/geographic distribution of Armillaria.
2. Materials and Methods
2.1. Fungal Specimens and Isolates
The investigation included 121 specimens loaned from six herbaria and these specimens were collected from 17 countries locating in Europe, North American, and Asia. Due to the absence of or limited Asian samples in previous phylogenetic studies, 105 strains isolated from sporocarps or rhizomorphs, covering 7 of 9 reported Armillaria species and 7 of 8 unnamed biological species in East Asia, were intensively collected in this study. In addition, 132 sequences from related strains/samples were downloaded from NCBI to represent species reported in Europe and North America. Since Desarmillaria species are the close relatives to Armillaria and recognized as Armillaria biological species, two Desarmillaria species were also included in this investigation. A total of 358 samples of Armillaria or Desarmillaria and related strains were included in the phylogenetic analyses. The detailed information of all samples used in this study is listed in the Supplementary Materials, Table S1.
For fresh sporocarps, spore prints were made on the same day of collection, stored in a cryogenic box, and brought back to the lab for single-spore isolation following the protocol described by Zhao et al. 1999 [38]. The collected rhizomorphs were first dipped in 50% ethanol for 15–20 s, and then sterilized in 20% hydrogen peroxide (H2O2) for 25–40 s depending on the thickness of the rhizomorphs. Both single spores and sterilized rhizomorphs were cultured on malt extract agar medium (12 g/L malt extract and 15 g/L Bacto Agar) amended with 2 mg/L benomyl and 100 mg/L streptomycin. All plates were incubated in darkness at 25 °C for 7–14 days to obtain isolated mycelia or rhizomorphs for DNA extraction.
2.2. DNA Extraction, PCR Amplification, and Sequencing
Genomic DNA was extracted from fresh cultured mycelia or rhizomorphs and sporocarp gills of dry specimens, at the junction of the stipe and pileus where spores are most possibly located, using a modified CTAB method [39]. Primers ITS1F and ITS4B [40] were used to amplify ITS region. Primers P-1 [41] and O-1 [42] were used to obtain the IGS-1 region. The partial TEF-1α gene was amplified with primer pairs EF595F/EF1160R [43]. Given that non-specific PCR products were obtained from some specimens with EF595F/EF1160R, a new primer pair, TEF-F (5′-GGCATCGAGGAGAGTCTTG-3′) and TEF-R (5′-TATCTCCAAGGACGGGCAGA-3′), was designed based on the TEF-1α gene of Armillaria. PCR reaction was performed in a total volume of 25 μL containing 2.5 μL of 10 × PCR buffer, 0.03 μL of dNTP mixture (10mM), 1 μL of each primer (10 pmol/μL), 0.15 μL of Taq polymerase (5 U/μL), and 1 μL of DNA template (50 ng/μL). PCR reactions were conducted in a ProFlexTM PCR system (Applied Biosystems, Foster City, CA, USA) under the following reaction conditions: denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C (for ITS) and 56 °C (for IGS-1 and TEF-1α) for 40 s and elongation at 72 °C for 1 min, a final elongation at 72 °C for 5 min. The purified products were sequenced in both directions on an ABI–3730 XL DNA Analyzer (Applied Biosystems, California).
2.3. Nucleotide Alignment and Data Matrices
The consensus DNA sequences were assembled using Seqman program in DNAstar (http://www.dnastar.com/t-seqmanpro.aspx, accessed on 12 July 2018). Alignments for each gene were generated using MAFFT 7 [44] and manually optimized in BioEdit 7.0.5 [45]. The three gene fragments were concatenated using Mesquite 3.31 [46].
Five data matrices were prepared. Dataset I (ITS+IGS-1+TEF-1α) including 358 samples was used to explore the phylogenetic relationship of all specimens and strains. Dataset I was also divided to three single-locus alignments to delimit phylogenetic species based on GCPSR [47]. Dataset II was set up to perform species delimitation using PTP model [36] and species validation in BPP program [48]. To balance the samples in each clade and satisfy the requirements in BPP program, 93 samples were chosen from Dataset I according to two following criteria: i) the number of samples in each well-supported clade ranged from 3 to 8 and strains from various geographic sites were chosen as much as possible; ii) samples with any gene locus absent were not included. Data III (ITS+IGS-1+TEF-1α) contained 270 concatenated sequences of samples in Gallica superclade and was used to perform neighbor-net phylogenetic network. Dataset IV (ITS+TEF-1α) was used to calibrate the most recent common ancestor (tMRCA) of the Armillaria clade, which included an extensive sampling of non-Armillaria outgroups from GenBank (Table S2). Dataset V (ITS+IGS-1+TEF-1α) was set up to run the second time of calibration, which included 22 Desarmillaria/Armillaria strains representing all phylogenetic species recognized in this study. In addition, the combined tree of Dataset V generated in BEAST was also employed for ancestral area reconstruction.
2.4. Phylogenetic Analyses and Species Delimitation
For Datasets I and II, Bayesian inference (BI) and Maximum Likelihood (ML) were performed using MrBayes 3.2 [49] and RAxML v.8 [50], respectively. The optimal evolutionary model was determined in MrModeltest [51] using the Akaike Information Criterion (AIC) for each locus. The selected substitution models for three loci were as follows: K81uf + Proportion Invariant + Gamma (K81uf + I + G) for ITS, General Time Reversible + I + G (GTR + I + G) for IGS-1, and Symmetrical model (SYM) + I + G for TEF-1α. The BI tree topology and posterior probabilities (PPs) were determined by a Markov Chain Monte Carlo (MCMC) algorithm of four chains with a random seed. Trees were sampled every 1000 generations with a burn in of the first 25% of trees. The MCMC analysis lasted until the average standard deviation of the split frequencies came below 0.01. The ML analysis was performed using RAxML, with 1000 bootstraps replicates, under the GTR-GAMMA model [50].
To delimit the species boundaries of Armillaria, three methods were applied. First, species were recognized by GCPSR following two steps [52]: (i) clades were genealogically concordant if they were present in >50% of the gene trees, and genealogically non-discordant if they were strongly supported (ML ≥ 70%; PP ≥ 0.95) in a single gene tree and not contradicted at or above this level of support in any other single gene tree; (ii) genetic differentiation and exhaustive subdivision criteria were applied [52]. Genetic differentiation required that lineages were well-differentiated, preventing minor terminal lineages from being recognized as phylogenetic species. Exhaustive subdivision required that all individuals were classified into phylogenetic species and no individuals were left unclassified. This technique involved tracing from the tip of the tree, and collapsing all lineages that were not subtended by an independent evolutionary lineage.
The second method used the PTP model, which stimulate the speciation or branching events in terms of number of substitutions based on a rooted phylogenetic tree [36]. The analysis was conducted on the web server for PTP (https://species.h-its.org/ptp/, accessed on 20 October 2020) using the RAxML tree estimated by Dataset II. Finally, a validation method, BPP, the reversible-jump Markov Chain Monte Carlo (rjMCMC) method [37], was used to test whether the candidate species suggested by GCPSR and PTP are reliable. This method tests alternative speciation hypotheses by collapsing or expanding nodes of the species tree using reversible-jump MCMC sampling in a fixed user-specified species tree and implemented through the software BPP 3.1 [53]. The program was run twice following the parameters below, species delimitation = 1, speciestree = 1, speciesmodelprior = 1, finetune ɛ = 1, usedata = 1 and cleandata = 0. The rjMCMC analyses consisted of 105 samples with a burn-in of 20%. The speciation event is validated if pp ≥ 0.95.
2.5. Pairwise Homoplasy Index Test
The concordance of gene genealogies can be used to evaluate the significance of gene flow between groups within an evolutionary time scale [54]. To determine the recombination level between every pair of clades in the Gallica superclade, a pairwise homoplasy index (PHI) test using the GCPSR recognized clade (based on Dataset II) was performed in SplitsTree v.4.14 [55]. A PHI value less than 0.05 indicated significant recombination.
2.6. Neighbor-net Network Analysis
To better present the phylogenetic relationships and geographical distribution of species in the Gallica superclade, all samples clustered in the Gallica superclade (Dataset III) were used to draw a neighbor-net phylogenetic network. The software SplitsTree v.4.14 [55] was applied using the default parameters with K2P-derived distances.
2.7. Molecular Dating Analysis
A two-step calibration procedure was used to estimate node ages using BEAST 2.4.5 [56]. We used BEAUTi to create XML files for Datasets IV. Two nodes were calibrated using fossil data: (i) the initial diversification of the marasmioid fungi (Marasmius alliaceus and Mycena pura) based on a 90 million-year-old (Ma) fossil Archaeomarasmius legetti [57]; (ii) the divergence between Hymenochaetaceae and Fomitopsidaceae based on a 125 Ma fossil, Quatsinoporites cranhamii [58]. For lognormal settings, the first calibration was logmean = 2.5, logstdev = 0.5, and offset = 90.0 and the second calibration was logmean = 2.0, logstdev = 0.5, and offset = 125.0. The concatenated nexus files of ITS and TEF-1α were created and uploaded into BEAUTi following the settings: GTR model, uncorrelated relaxed clock with lognormal rate distribution, and the birth-death prior set. Three independent runs of 107 MCMC steps with random starting seeds were carried out, with sampling at every 1000 generations, following a burn-in of the initial 10% cycles. The convergence of runs was checked using Tracer with an effective sample size (ESS) > 200. The maximum clade credibility (MCC) tree was summarized using TreeAnnotator [59] and visualized in FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 15 July 2019) to obtain the means and 95% higher posterior densities (HPDs).
2.8. Ancestral Area Reconstruction
To understand the biogeography of Armillaria, an ancestral area reconstruction analysis was inferred using the Bayesian Binary MCMC (BBM) analysis [60] and Dispersal-Extinction-Cladogenesis (DEC) model [61] and as implemented in the software of RASP 3.1 [62]. Based on the tectonic history of Laurasia, the geographic distributions for Armillaria were delineated into three areas: eastern Asia (A), Europe (B), and North America (C). Each taxon in our dataset was assigned to an area based on its current known distribution range. For the DEC model, the dispersal probability between A and B was unconstrained and the dispersal probabilities of the other two pairs, A←→C and B←→C, were constrained to 0.1 as summarized by previous studies [63]. The BBM analysis was conducted by setting the generation to 10 million with a burn-in of 20% and other parameters used default options.
3. Results
3.1. Phylogenetic Analyses and Species Delimitation
The concatenated sequences of Dataset I (ITS+IGS-1+TEF-1α) were 2010 bp in length (with gap), containing 821 polymorphic sites (320 from IGS-1, 280 from TEF-1α, and 221 from ITS). The nucleotide diversity (Pi) of TEF-1α and IGS-1 was 5.0 × 10−2 and 4.9 × 10−2, relatively higher than that of ITS (Pi = 1.6 × 10−2). The ML and BI analyses yielded identical tree topology with six well-supported clades. The concatenated tree inferred from ML analysis is shown in Figure 1 and the single gene trees are shown in Figure S1. According to the “superclade” identified by Klopfenstein et al. 2017 [27], 226 samples sequenced in our study were assigned to the Gallica superclade (185 samples), Solidipes/Ostoyae superclade (26 samples), Mellea superclade (12 samples), and Desarmillaria genus (3 samples). Three Chinese strains isolated from rhizomorph samples associated with Gastrodia elata in the natural field clustered in an additional clade, Clade III. Since Desarmillaria species do not produce rhizomorphs in nature, Clade III could represent a new superclade in Armillaria. Whether Clade III is the most ancient Armillaria clade needs more samples together with southern hemisphere species to verify. These strains were not included in the following species delimitation due to the absence of IGS-1 sequences.
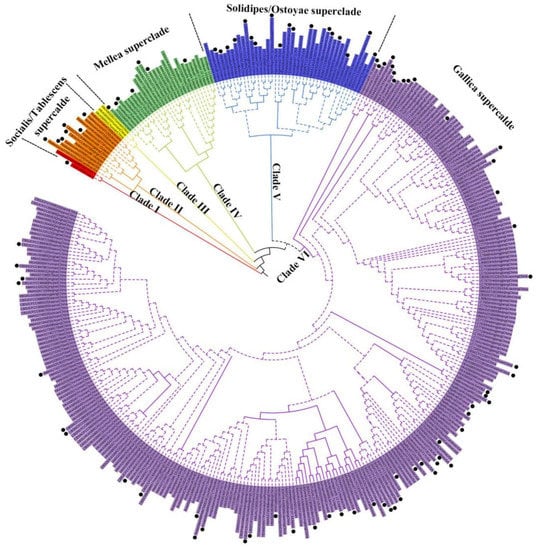
Figure 1.
Phylogenetic tree of 358 Armillaria and Desarmillaria samples inferred from maximum likelihood and Bayesian analyses based on Dataset I (ITS+IGS-1+TEF-1α). Only bootstraps (LB) over 70% and Bayesian posterior probabilities (PPs) over 0.95 are shown on the branches. The samples labeled with black dots were chosen for the species delimitation analysis presented in Figure 2.
For Dataset II (93 representative samples), all species delimitation methods yielded a consistent result that previously defined D. tabescens and A. mellea, where each consisted of three strong supported clades geographically limited to East Asia, North America, and Europe. Here, we recorded D. tabescens as D. tabescens-EA, D. tabescens-NA, and D. tabescens-EU. Similarly, A. mellea herein was recorded as A. mellea-EA, A. mellea-NA, and A. mellea-EU. In the Ostoyae/Solidipes superclade, due to limited samples, the phylogenetic relationships among representatives were not completely resolved. Armillaria ostoyae collected from Japan and European countries were clustered in two separate clades, suggesting that A. ostoyae was not monophyletic (Figure 2). Three CBS M (CFCC 88644, CFCC 80932, CFCC 8884) specimens, previously defined as A. borealis due to their mating compatibility test with European A. borealis [21], however, were not clustered together but clearly phylogenetically separated by two A. ostoyae clades (Figure 2). In the Gallica superclade, 11 candidate taxonomic units (according to 11 clades in Figure 2) were designated based on the bootstrap or posterior probability values according to GCPSR. Whereas, PTP analysis only recognized three species, i.e., A. nabsnona (clade 11), Phylogenetic species I (PS I, i.e., clade 10, previously defined as Nag. E) [64], and PS II (clade I to clade 9) (Figure 2 and Figure S2).

Figure 2.
Species delimitation of Armillaria and Desarmillaria based on a phylogenetic constructed using Dataset II (ITS+IGS-1+TEF-1α)) consisting of 93 representative strains. Only maximum likelihood bootstraps (LBs) over 70% and Bayesian posterior probabilities (PPs) over 0.95 are shown on the branches.
In order to test the discrepancy of the hypothesized species inferred from PTP and GCPSR, additional analysis was performed by running BPP. A dataset was composed of strains of three potential species (A. nabsnona, PS I, and PS II) and three reference species, three cryptic species in the Mellea superclade (A. mellea-EU, A. mellea-EA, and A. mellea-NA) [28]. Four combinations with a varied population size and divergence rates were analyzed. Both analyses with large ancestral population size (Gθs: 1, 10) supported four reference species and three species of the Gallica superclade delimited in PTP based on the GCPSR result; clade 10 and clade 11, corresponding to A. nabsnona and PS I, were still strongly supported while several potential species in clade 1 to clade 9 were not supported to be distinct species due to low posterior possibility values (<0.95) (Table 1). Both PTP and BPP supported that the Gallica superclade contained three phylogenetic species. Previously defined “A. nabsnona” and “Nag. E” were confirmed to be two well-supported species and recorded as A. nabsnona and PS I in this study while previously defined “A. gallica”, “A. cepistipes”, “A. calvescens”, “A. altimontana”, and “A. sinapina” and seven Chinese Biological Species” (CBS C, CBS F, CBS H, CBS J, CBS L, CBS N, and CBS O) were collectively recognized as a single species and recorded herein as PS II.

Table 1.
Results from BPP analyses for the Gallica superclade assuming 3-species or 11-species models.
When applying PHI tests, no significant recombination was detected between pairs of A. nabsnona, PS I, and PS II, but significant recombination was detected among different clade-pairs within PS II (Table 2), which indicated that the reproduction isolation had formed among A. nabsnona, PS I, and PS II but not formed completely within PS II.
Table 2.
PHI test (above diagonal) and compatible test (below diagonal) of phylogenetic clades within the Gallica superclade.
3.2. Phylogenetic Relationships in PS II
The neighbor-net network revealed the presence of divergent lineages in the Gallica superclade. The Gallica superclade was divided into multiple lineages as A. nabosnoa and PS I (Nag. E) were recognized as distinct distant lineages (Figure 3). Within PSII, previously unnamed six CBS “species” (CBS C, CBS H, CBS J, CBS L, CBS N, and CBS O), together with many Chinese samples were grouped as lineage 1. Among the 87 samples in this lineage, 81 (93%) samples were collected from Qinghai-Tibet Plateau and adjoining areas covering Tibet, Qinghai, Xinjiang, Sichuan, Yunnan, Shaanxi, and Guizhou Provinces (Table S1). Therefore, we recorded this lineage as the Chinese endemic lineage. Lineage 2 contained North America-centric samples, including two North America-specific species, A. altimontana and A. calvescens, as well as the specimens of A. gallica in North America. Lineage 3 comprised CBS A, CBS F, A. cepistipes, and A. sinapina. The divergence within A. cepistipes and A. sinapina could indicate the existence of geographical divergence. Armillaria sinapina and CBS A were clustered together, but samples from North America and East Asia formed two sister clades. A. cepistipes collected from North America were clustered in two distinct clades and CBS F were grouped to two distinct clades, one with A. cepistipes from Europe and North America and another with only Chinese samples. In lineage 4, A. gallica collected from East Asia and Europe as well as CBS B were included.
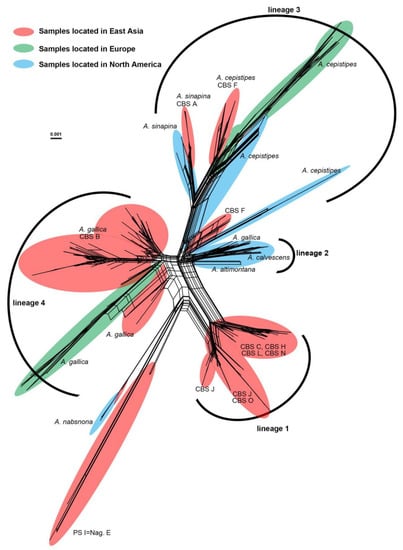
Figure 3.
A neighbor-net phylogenetic network based on partial sequences of ITS, IGS-1, and TEF-1α from 270 Armillaria samples in the Gallica superclade created by SplitsTree v4.14.4 with K2P distance. The names of previously defined species were listed nearby the lineages. Samples located in East Asia, Europe, and North America were highlighted in red, green, and blue, respectively. Detailed information of 270 Armillaria samples is described in Table S1.
3.3. Estimation of Divergence Time
The first molecular dating with two fossils suggested that the most recent common ancestor (tMRCA) of Armillaria and Desarmillaria was estimated at early Miocene (21.8 Mya, 95% HPD: 13.1–32.6 Mya, Figure 4). The second calibration showed the divergence date of the Solidipes/Ostoyae superclade and Gallica superclade was estimated at 15.7 Mya (95% HPD: 3.5–36.2 Mya) (node 3 in Table 3, Figure 4). The diversification of the Gallica superclade was estimated to have occurred at 9.1 Mya (95% HPD 21.4–1.2 Mya) (node 5 in Table 3, Figure 4). The PS I separated from its tMRCA during late Miocene to Pleistocene (6.9 Mya, 95% HPD:1.1–16.2 Mya) (node 6 in Table 3, Figure 4). Four lineages of PS II diverged during late Miocene to Pleistocene (95% HPD: 10.1–0.6 Mya) (node 8 in Table 3, Figure 4).

Figure 4.
Chronogram of Armillaria from the northern hemisphere. The time-scale is set to the mean divergence dates produced in BEAST. The most recent common ancestor (tMRCA) of Armillaria and Desarmillaria (node 1) was estimated based on Dataset IV and its chronogram is shown in Figure S3. Numbered nodes refer to mean divergence dates, with their 95% HPD and ancestral state provided in Table 3. The pie chart in each node indicates the possible ancestral distributions inferred from Bayesian Binary MCMC analysis (BBM) implemented in RASP. Red branches identify dispersal events inferred by RASP based on the Dispersal-Extinction-Cladogenesis (DEC) model.
Table 3.
Divergence time estimates of BEAST analyses for internal nodes of northern hemisphere Armillaria, with results of ancestral range estimation using the BBM and DEC models.
3.4. Ancestral Areas of Armillaria
The most probable ancestral areas for internal nodes of Armillaria are not completely consistent in the BBM and DEC analyses. In the BBM analyses, most nodes were inferred as a single region of origin, such as East Asia or North America, and East Asia was the most probable ancestral area for Armillaria. While in the DEC analyses, multi-regional origins covering East Asia, Europe, and North America were inferred in several nodes (Table 3), but the relative probabilities of the ancestral area for some nodes are very low (nodes 1 and 3). Some nodes were consistently supported by BBM and DEC analyses, such as the ancestral areas of A. nabosnona (node 5), PS I+PS II (node 6), PS II (node 7), and lineage 1 in PS II, which were all suggested to originate from East Asia.
4. Discussion
Based on phylogenetic analyses and multiple species delimitation methods, our results supported the monophyly of “biological species” (“BS”) D. tabescens and A. mellea, but within each of these two “BS”, allopatric speciation has been detected. For instance, “BS” A. mellea was composed of three distinct phylogenetic species geographically limited to North America (A. mellea-NA), East Asia (A. mellea-EA), and Europe (A. mellea-EU) (Figure 2). “BS” D. tabescens also presented the same geographic grouping (NA, EA, and EU). Whereas in the Gallica superclade, 11 “biological species” were revealed to represent only 3 phylogenetic species (Figure 2, Table 1), i.e., the previously recognized CBS C, CBS H, CBS J, CBS L, CBS N, and CBS O [21], although clustered together but failed to be distinguished from each other. In the Solidipes/Ostoyae superclade, CBS D and “BS” A. ostoyae mixed together but distributed in two non-adjacent phylogenetic clades, with each clade consisting of both CBS D and “BS” A. ostoyae. This interlaced distribution was also shown in a previous study [26]. Therefore, the previously established relation between “BS” A. ostoyae and CBS D should be challenged [69], because “BS” A. ostoyae is not monophyletic, dividing into at least two distinct lineages (Figure 2). The pronounced discrepancies between “biological species” and phylogenetic grouping suggested that biological species recognition did not reflect the natural evolutionary relationships within Armillaria. Besides its ambiguous criterion and complicated operability as criticized by many mycologists [70], biological recognition is particularly inadequate for Armillaria species. In previous studies, the sole detection of hyphal fusion and diploidy in “compatibility tests” was improperly considered as the evidence of mating success in Armillaria [20]. Additionally, hyphal fusion can occur at the interspecific level, as reported in smut fungi from different families [70]. Mating success requires not only hyphal fusion but also fertile offspring production to be achieved. Previous compatibility tests did not check whether the fused diploid could produce fertile offspring. A similar example is Neurospora tetrasperma complex [71]. Nine phylogenetic species were recognized following PSR. If using the traditional broad biological species recognition, hyphal fusion, these nine phylogenetic species constituted a single biological species. However, when examining the reproductive success, such as the viability and fertility of offspring, BSR also supports the nine species delimitation. Nevertheless, it is difficult to measure the fertility of offspring in macrofungi, e.g., Desarmillaria and Armilllaria. So far, the in vitro fruiting system of Armillaria was only reported in the “BS” A. ostoyae and A. mellea-NA lineage [72,73], while hyphae-fused Armillaria “biological species” have been widely and mistakenly recognized. Speciation is a dynamic and ongoing evolutionary process, through which a species arises. The inconsistency among species concepts could be caused by different phenotypic and/or genotypic features used in different species concepts/recognition [74]. With more and more genome data becoming available, genome sequence-based classification and identification has appealed recently [74,75]. The validity of multi-geographic limited species in Tabescens and Mellea superclades should be evaluated with genome data in the future. Our estimates of the tMRCA of Armillaria in the northern hemisphere suggest it arose at 21.8 (32.6–13.1, 95% HPD) Mya, overlapped with a previous estimate (43.4–17.7 Mya, 95% HPD) based on a global phylogeny of Armillaria [17], even though a different set of loci was used. During this time period, Boreotropical flora (56–33.9 Mya) [76] was establishing (if adopting Koch’s estimation) or had resulted in a global shift of vegetation in the northern hemisphere. Many large and fast-growing trees, such as alpine coniferous deciduous forests, had emerged [77]. These plants were frequently recorded as common substrates of Armillaria spp. [21,66,78], which may facilitate long-distance transcontinental dispersal.
In our case, allopatric speciation due to geographic isolation was shown in “BS” A. mellea and D. tabescens. Three clades corresponding to their geographic locations in Asia, Europe, and North America were revealed in A. mellea. These geographic clades were well supported and recognized as distinct species based on data analysis (Figure 2). The North America clade can be further divided into eastern and western clades based on microsatellite loci [79]. These geographic clades were accepted as cryptic species in the Mellea superclade recently [26]. Besides, A. mellea was also reported from Africa [80], Mexico [81], and Iran [82]. A more comprehensive phylogeny with samples from different countries should be constructed to test the existence of cryptic species in the Mellea superclade. Similarly, three continental-specific clades of D. tabescens were recognized as distinct species and highly supported by the molecular delimitation methods. The DNA variation among three continental specific clades of D. tabescens is even greater than the ever proposed species in the Gallica superclade. Strains from East Asia, Europe, and North America were considered as conspecific based on incongruent placement on two single gene trees (IGS-1 and TEF-1α) [26]. Nevertheless, a recent five gene phylogeny (28S, TEF-1α, rpb2, act, and gpd) supported the phylogenetic variation between D. tabescens-NA and D. tabescens from Eurasia and introduced D. tabescens-NA as a new species, D. caespitosa, due to significant morphological differences including wider basidiospores, narrower cheilocystidia, and caulocystidia [29]. Our results suggested that not only D. tabescens-NA, but also D. tabescens-EA could present a new species. Our results supported a recent speculation that even more crytic species existed in the Tabescens superclade estimated by ITS1 or ITS2 from public databases [83]. Previous studies indicated several factors could contribute to allopatric speciation, such as genetic drift in spatial populations and natural selection [84,85]. With more available Armillaria genome sequences [86], the mechanisms of allopatric speciation and adaptations in Mellea and Tabescens superclades should be analyzed at the genomic level in future study.
The first species separated from the crown node of the Gallica superclade was A. nabsnona, which showed a disjunctive distribution in East Asia and the west coast of North America [30,87]. The divergence time of A. nabsnona was around 9.1 Mya, earlier than the opening of the Bering Strait (node 5: Table 3 and Figure 4). The long-distance transcontinental dispersal, incremental dispersal over land, and vicariance may have contributed to the disjunctive distribution of A. nabsnona. With new sampling in this study, PS I, previously recorded as Nag. E only reported from Japan, showed a broader distribution including one specimen from Tibet, China (Figure 2, Table S1).
In the Gallica superclade, multiple delimitation methods recognized several previously defined “biological species” as a single phylogenetic species, PS II (Figure 2), regardless of their variability in host range and morphologies. However, the phylogenetic network indicated lineage divergence in PS II, which is experiencing an ongoing speciation process. Armillaia samples from Qing-Tibet Plateau formed a distinct lineage (lineage 1) and molecular dating showed that this lineage arose at 2.2 Mya (6.2–0.1 Mya, 95% HPD) (Figure 4), in concurrence with Pliocene uplift of the Northern Tibetan Plateau (since 4.5 Mya) [88]. We hypothesized that the appearance of this endemic Armillaria lineage was driven by the uplift of the Northern Tibetan Plateau. It has been well documented that the Pliocene uplift of Himalaya resulted in critical local climate change at around 5 Mya [89,90]. High species diversity and potential origins of many well-known species in different groups from the Himalaya areas have also been reported, such as Saccharomyces cerevisiae in fungi [91], Quercus spp. in plants [92], and Coelotine spp. spiders in animals [93]. Therefore, the divergence of lineage 1 could be a consequence of adaptive evolution and geographic isolation. Neither recombination tests of the Parsimony Tree Length Permutation Test (PTLPT) nor linkage disequilibrium (rd) showed signal of recombination within lineage 1 (Figure S4), suggesting that lineage 1 is expanding its population size via the clonal reproductive mode. Several commercial strains (HKAS 86556, HKAS 86,557, and HKAS 86543), widely used in the growth of Gastrodia elata (a traditional medicinal plant), clustered in lineage 1. Frequent introduction of these commercial plants may have contributed to its clonal expansion.
Lineage 3 and lineage 4 comprised clades of different geographic origins (Figure 3). The tMRCA (node 9) of lineage 2, lineage 3, and lineage 4 was estimated at 4.2 Mya (Figure 4, Table 3), later than the opening of the Bering Strait (5.5–5.4 Mya) [94]. Therefore, the vicariance event, such as the opening of the Bering Strait, could have driven the divergence of lineages located in North America and East Asia. In lineage 3, samples located in North America and East Asia were genetically separated (Figure 3) and a similar genetic distinction was also revealed in other wood-decay fungi, i.e., Coniferiporia sulphurascens [95]. Although Armillaria could disperse over long distances via basidiospores [16], it is limited by the Pacific Ocean [96]. One possible explanation was that the common ancestors of continental samples in lineage 3 spread via the Bering land bridge to North America to attain a transcontinental distribution and subsequently diverged when vicariance events occurred.
A previous study roughly suggested Eurasia was the most probable ancestral area for Armillaria [17]. In this study, with much more extensive samplings in the northern hemisphere, the source of Armillaria origin was narrowed to East Asia with 88% probability in BBM analysis (Figure 4 and Table 3). Although the DEC analysis suggested that the Armillaria genus was probably a multi-range origin, the probability was too low (ABC, 33%, Table 3) to be accepted. The most ancient clade in Armillaria is Clade III (Figure 1), which included three samples representing an undescribed phylogenetic species so far known only from China. The earliest lineage diverged from Clade III is A. mellea-EA, an East Asian geographically limited species, which included both heterothallic and homothallic reproductive systems [12]. The phylogeny and neighbor-net analysis of northern Armillaria spp. revealed that samples from East Asia distributed in every major evolutionary clade or lineage, indicating a diversity center with high species richness and genetic diversity of Armillaria in East Asia. As estimated by Koch et al. [83], Eastern Asia represents the biogeographic region with the highest species richness. In a detailed analysis of the largest evolutionary clade, the Gallica superclade, both BBM and DEC analyses supported an East Asian origin (Figure 4 and Table 3). Therefore, it is reasonable to speculate that East Asia is the origin of the Armillaria genus in the northern hemisphere, as well as the origin of most major evolutionary lineages in Armillaria.
5. Conclusions
In summary, the phylogenetic species approach based on rRNA and TEF-1α indicated the presence of cryptic species and lineages within Armillaria, giving more natural species delimitation than biological species recognition. Geographic isolation has been implicated to be a key determinant in the speciation and lineage divergence. Armillaria was inferred to originate from East Asia at Early Miocene and the basal clade had evolved to three distinct species after dispersing to other continents. However, in the Gallica clade, most previously defined species could represent lineages of PS II, a super species experiencing an ongoing speciation process. Divergence time estimation suggested that both the transcontinental dispersal within the boreal floristic kingdom and vicariance events during early Pliocene could have led to a disjunctive distribution and lineage divergence of Armillaria species in the Gallica superclade.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jof7121088/s1, Figure S1: Single locus phylogenetic analyses of 358 Desarmillaria and Armillaria strains used in this study based on ITS (a), IGS-1 (b) and TEF-1α (c). Only maximum likelihood bootstraps (LB) over 70% and Bayesian posterior probabilities (PP) over 0.95 were showed on the branches, Figure S2: Results of the PTP analysis based on the BI and ML topologies. Putative species clusters are indicated using transitions between blue and red branches. Figure S3: Molecular dating of tMRCA of Armillaria and Desarmillaria with 27 reference strains representing major clades of Agaricomycetes, Dacrymycetes and Tremellomycetes. The time-scale is set to the mean divergence dates produced in BEAST v. 2.4.5, Figure S4: Recombination test of Armillaria Chinese specific population of PS II based on linkage disequilibrium (rd) and parsimony tree lengths (PTLPT). Ranges of PTLPT and rd were conducted from 1000 randomized permutations using Multilocus v. 1.3, Table S1: Detailed information of 358 Armillaria and Desarmillaria samples used in this study, Table S2: Species and their sequences accession number used for molecular clock analysis.
Author Contributions
Conceptualization, J.L. and C.K.M.T.; methodology, J.L.; software, J.L.; validation, J.L.; formal analysis, J.L.; investigation, J.L.; resources, J.L., P.Z., Z.Z. and Z.Y.; data curation, J.L.; writing—original draft preparation, J.L.; writing—review and editing, J.L., L.C., L.P. and C.K.M.T.; visualization, J.L.; supervision. L.P. and C.K.M.T.; project administration, J.L.; funding acquisition, J.L. and C.K.M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (NFSC) (grant number 31600405 and 31972210) and Biological Resources Program, Chinese Academy of Sciences (grant number KFJ-BRP-009). Clement K.M. Tsui is grateful to CAS President’s International Fellowship Initiative for the award of a visiting scientist fellowship for scientific exchange (grant number 2019VBC0006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data of this study are included in the article or Supplementary Materials.
Acknowledgments
The authors are grateful to following herbaria for providing us with Armillaria/Desarmillaria samples: Herbarium of Mycology, Jilin Agricultural University (HMJAU), Herbarium of Mycology, Chinese Academy of Science (HMAS), University of Washington Herbarium (WTU), University of Tennessee Herbarium (TENN), University of Minnesota Herbarium (MIN).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berbee, M.L.; Taylor, J.W. Dating the molecular clock in fungi-how close are we? Fungal Biol. Rev. 2010, 24, 1–16. [Google Scholar] [CrossRef]
- Tharreau, D.; Fudal, I.; Andriantsimialona, D.; Santoso, U.D.; Fournier, E.; Lebrun, M.H.; Nottéghem, J.L. World population structure and migration of the rice blast fungus, Magnaporthe oryzae. In Advances in Genetics, Genomics and Control of Rice Blast Disease; Wang, G.L., Valent, B., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 209–215. [Google Scholar]
- Saleh, D.; Milazzo, J.; Adreit, H.; Fournier, E.; Tharreau, D. South-East Asia is the center of origin, diversity and dispersion of the rice blast fungus, Magnaporthe oryzae. New Phytol. 2014, 201, 1440–1456. [Google Scholar] [CrossRef]
- Manos, P.S.; Stanford, A.M. The Historical Biogeography of Fagaceae: Tracking the Tertiary History of Temperate and Subtropical Forests of the Northern Hemisphere. Int. J. Plant Sci. 2001, 162, S77–S93. [Google Scholar] [CrossRef]
- Taylor, J.W.; Berbee, M.L. Dating divergences in the Fungal Tree of Life: Review and new analyses. Mycologia 2006, 98, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Percy, D.M.; Garver, A.M.; Wagner, W.L.; James, H.; Cunningham, C.; Miller, S.; Fleischer, R.C. Progressive island colonization and ancient origin of Hawaiian Metrosideros (Myrtaceae). Proc. R. Soc. B Boil. Sci. 2008, 275, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Han, L.L.; Feng, B.; Wu, G.; Halling, R.E.; Buyck, B.; Yorou, N.S.; Wbika, S.T.N.; Yang, Z.L. African origin and global distribution patterns: Evidence inferred from phylogenetic and biogeographical analyses of ectomycorrhizal fungal genus Strobilomyces. J. Biogeogr. 2018, 45, 201–212. [Google Scholar] [CrossRef]
- Cai, Q.; Tulloss, R.E.; Tang, L.P.; Tolgor, B.; Zhang, P.; Chen, Z.H.; Yang, Z.L. Multi-locus phylogeny of lethal amanitas: Implications for species diversity and historical biogeography. BMC Evol. Biol. 2014, 14, 143. [Google Scholar] [CrossRef]
- Song, J.; Cui, B.K. Phylogeny, divergence time and historical biogeography of Laetiporus (Basidiomycota, Polyporales). BMC Evol. Biol. 2017, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Truong, C.; Sanchez-Ramirez, S.; Kuhar, F.; Kaplan, Z.; Smith, M.E. The Gondwanan connection-Southern temperate Amanita lineages and the description of the first sequestrate species from the Americas. Fungal Biol. 2017, 121, 638–651. [Google Scholar] [CrossRef]
- Boluda, C.G.; Rico, V.J.; Naciri, Y.; Hawksworth, D.L.; Scheidegger, C. Phylogeographic reconstructions can be biased by ancestral shared alleles: The case of the polymorphic lichen Bryoria fuscescens in Europe and North Africa. Mol. Ecol. 2021, 30, 4845–4865. [Google Scholar] [CrossRef]
- Baumgartner, K.; Coetzee, M.; Hoffmeister, D. Secrets of the subterranean pathosystem of Armillaria. Mol. Plant Pathol. 2011, 12, 515–534. [Google Scholar] [CrossRef]
- Smith, M.; Bruhn, J.N.; Anderson, J.B. The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nat. Cell Biol. 1992, 356, 428–431. [Google Scholar] [CrossRef]
- Hobbs, C. Medicinal Mushrooms: An Exploration of Tradition, Healing and Culture; Botanica Press: Summertown, TN, USA, 2003. [Google Scholar]
- Kikuchi, G.; Yamaji, H. Identification of Armillaria species associated with Polyporus umbellatus using ITS sequences of nuclear ribosomal DNA. Mycoscience 2010, 51, 366–372. [Google Scholar] [CrossRef]
- Coetzee, M.P.A.; Bloomer, P.; Wingfield, M.J.; Wingfield, B. Paleogene Radiation of a Plant Pathogenic Mushroom. PLoS ONE 2011, 6, e28545. [Google Scholar] [CrossRef]
- Koch, R.A.; Wilson, A.W.; Séné, O.; Henkel, T.W.; Aime, M.C. Resolved phylogeny and biogeography of the root pathogen Armillaria and its gasteroid relative, Guyanagaster. BMC Evol. Biol. 2017, 17, 1–16. [Google Scholar] [CrossRef]
- Watling, R.; Kile, G.; Gregory, N.M. The genus Armillaria-nomenclature, typification, the identity of Armillaria mellea and species differentiation. Trans. Br. Mycol. Soc. 1982, 78, 271–285. [Google Scholar] [CrossRef]
- Volk, T.J.; Burdsall, H.H. A Nomenclatural Study of Armillaria and Armillariella Species: Basidiomycotina, Tricholomataceae; Fungiflora Press: Oslo, Norway, 1995. [Google Scholar]
- Korhonen, K. Interfertility and clonal size in the Armillaria mellea complex. Karstenia 1978, 18, 31–42. [Google Scholar] [CrossRef]
- Qin, G.F.; Zhao, J.; Korhonen, K. A study on intersterility groups of Armillaria in China. Mycologia 2007, 99, 430–441. [Google Scholar] [CrossRef]
- Nixon, K.C.; Wheeler, Q.D. An Amplification of the Phylogenetic Species Concept. Cladistics 1990, 6, 211–223. [Google Scholar] [CrossRef]
- O’Donnell, K. Molecular phylogeny of the Nectria haematococca–Fusarium solani species complex. Mycologia 2000, 92, 919–938. [Google Scholar] [CrossRef]
- Maryani, N.; Lombard, L.; Poerba, Y.; Subandiyah, S.; Crous, P.; Kema, G. Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud. Mycol. 2019, 92, 155–194. [Google Scholar] [CrossRef]
- Weir, B.; Johnston, P.; Damm, U. The Colletotrichum gloeosporioides species complex. Stud. Mycol. 2012, 73, 115–180. [Google Scholar] [CrossRef]
- Coetzee, M.P.; Wingfield, B.; Zhao, J.; van Coller, S.J.; Wingfield, M.J. Phylogenetic relationships among biological species of Armillaria from China. Mycoscience 2015, 56, 530–541. [Google Scholar] [CrossRef]
- Klopfenstein, N.B.; Stewart, J.E.; Ota, Y.; Hanna, J.W.; Richardson, B.A.; Ross-Davis, A.L.; Elías-Román, R.D.; Korhonen, K.; Keča, N.; Iturritxa, E.; et al. Insights into the phylogeny of Northern Hemisphere Armillaria: Neighbor-net and Bayesian analyses of translation elongation factor 1-α gene sequences. Mycologia 2017, 109, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, M.P.; Wingfield, B.D.; Wingfield, M.J. Armillaria Root-Rot Pathogens: Species Boundaries and Global Distribution. Pathogens 2018, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Antonín, V.; Stewart, J.E.; Ortiz, R.M.; Kim, M.-S.; Bonello, P.; Tomšovský, M.; Klopfenstein, N.B. Desarmillaria caespitosa, a North American vicariant of D. tabescens. Mycologia 2021, 113, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Volk, T.J.; Burdsall, H.H.; Banik, M.T. Armillaria nabsnona, a New Species from Western North America. Mycologia 1996, 88, 484–491. [Google Scholar] [CrossRef]
- Berube, J.A.; Dessureault, M. Morphological studies of the Armillaria mellea complex: Two new species, A. gemina and A. calvescens. Mycologia 1989, 81, 216–225. [Google Scholar] [CrossRef]
- Brazee, N.J.; Ortiz-Santana, B.; Banik, M.T.; Lindner, D.L. Armillaria altimontana, a new species from the western interior of North America. Mycologia 2012, 104, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Sung, J.M.; Igarashi, T. Biological species and morphological characteristics of Armillaria mellea complex in Hokkaido: A. sinapina and two new species, A. jezoensis and A. singula. Mycoscience 1994, 35, 39–47. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.; Gómez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Bayesian species identification under the multispecies coalescent provides significant improvements to DNA barcoding analyses. Mol. Ecol. 2017, 26, 3028–3036. [Google Scholar] [CrossRef]
- Zhao, J.; Tian, S.; Wang, Y.; Qin, G. A method of single spore isolation and culture for Armillaria genetic test. Microbiology 1999, 26, 207–209. [Google Scholar]
- Coetzee, M.P.A.; Wingfield, B.D.; Harrington, T.C.; Dalevi, D.; Coutinho, T.A.; Wingfield, M.J. Geographical diversity of Ar-millaria mellea s. s. based on phylogenetic analysis. Mycologia 2000, 92, 105–113. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hsiau, T.W. The Taxonomy and Phylogeny of the Mycangial Fungi from Dendroctonus brevicomis and D. frontalis (Coleoptera: Scolytidae). Ph.D. Thesis, Lowa State university, Ames, IA, USA, 1996. [Google Scholar]
- Duchesne, L.C.; Anderson, J.B. Location and direction of transcription of the 5S rRNA gene in Armillaria. Mycol. Res. 1990, 94, 266–269. [Google Scholar] [CrossRef]
- Kauserud, H.; Schumacher, T. Outcrossing or inbreeding: DNA markers provide evidence for type of reproductive mode in Phellinus nigrolimitatus (Basidiomycota). Mycol. Res. 2001, 105, 676–683. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D. MAFFT multiple sequence alignment software version improvements in performance and usability. Mol. Biol. Evo. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Alzohairy, A.M. Bioedit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Version 3.31. 2017. Available online: https://www.mesquiteproject.org (accessed on 6 November 2021).
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in Fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 2010, 107, 9264–9269. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Hoehna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Nylander, J.A.A. MrModeltest 2.3 README, 22 May 2008. Syst. Zool. 2008, 3–4. [Google Scholar]
- Dettman, J.R.; Jacobson, D.J.; Taylor, J.W. Multilocus sequence data reveal extensive phylogenetic species diversity within the Neurospora discreta complex. Mycologia 2006, 98, 436–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, Z. The BPP program for species tree estimation and species delimitation. Curr. Zoöl. 2015, 61, 854–865. [Google Scholar] [CrossRef]
- Quaedvlieg, W.; Binder, M.; Groenewald, J.; Summerell, B.; Carnegie, A.; Burgess, T.; Crous, P. Introducing the Consolidated Species Concept to resolve species in the Teratosphaeriaceae. Persoonia, Mol. Phylogeny Evol. Fungi 2014, 33, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Kloepper, T.; Bryant, D. SplitsTree 4.0-Computation of phylogenetic trees and networks. Bioinformatics 2008, 14, 68–73. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.; Grimaldi, D.; Donoghue, M. Fossil mushrooms from Miocene and Cretaceous ambers and the evolution of Homobasidiomycetes. Am J. Bot. 1997, 84, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.Y.; Currah, R.S.; Stockey, R.A. Cretaceous and Eocene poroid hymenophores from Vancouver Island, British Co-lumbia. Mycologia 2004, 96, 180–186. [Google Scholar] [CrossRef]
- Drummond, R.B.A.J.; Rambaut, A.A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2017, 7, 214. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Ree, R.H.; Smith, S. Maximum Likelihood Inference of Geographic Range Evolution by Dispersal, Local Extinction, and Cladogenesis. Syst. Biol. 2008, 57, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Harris, A.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenetics Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Tiffney, B.H. The Eocene North Atlantic land bridge:its importance in Tertiary and modern phytogeography of the Northern Hemisphere. J. Arnold Arboretum. 1985, 66, 243–273. [Google Scholar] [CrossRef]
- Nagasawa, E. Taxonomic reassessment of Armillaria mellea in Japan. Report for a Grant-in-Aid for Scientific Research. 1991. 63560155. Available online: https://kaken.nii.ac.jp/en/grant/KAKENHI-PROJECT-63560155/ (accessed on 4 November 2021).
- Anderson, J.B.; Ullrich, R.C. Biological Species of Armillaria mellea in North America. Mycologia 1979, 71, 402. [Google Scholar] [CrossRef]
- Anderson, J.B.; Korhonen, K.; Ullrich, R.C. Relationships between European and North American biological species of Armillaria mellea. Exp. Mycol. 1980, 4, 78–86. [Google Scholar] [CrossRef]
- Banik, M.T.; Burdsall, H.H. Assessment of Compatibility among Armillaria cepistipes, A. sinapina, and North American Biological Species X and XI, Using Culture Morphology and Molecular Biology. Mycologia 1998, 90, 798. [Google Scholar] [CrossRef]
- Ota, Y.; Matsushita, N.; Nagasawa, E.; Terashita, T.; Fukuda, K.; Suzuki, K. Biological Species of Armillaria in Japan. Plant Dis. 1998, 82, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Wang, H.C.; Xue, W.Q.; Zhao, J.; Yang, Z.L. Phylogenetic analyses of Armillaria reveal at least 15 phylogenetic Lineages in China, seven of which are associated with cultivated Gastrodia elata. PLoS ONE 2016, 11, e0154794. [Google Scholar] [CrossRef]
- Kellner, R.; Vollmeister, E.; Feldbrügge, M.; Begerow, D. Interspecific sex in grass smuts and the genetic diversity of their pheromone-receptor System. PLoS Genet. 2011, 7, e1002436. [Google Scholar] [CrossRef] [PubMed]
- Menkis, A.; Bastiaans, E.; Jacobson, D.J.; Johannesson, H. Phylogenetic and biological species diversity within the Neurospora tetrasperma complex. J. Evol. Biol. 2009, 22, 1923–1936. [Google Scholar] [CrossRef]
- Zhuang, Y.; Zhao, J.; Qin, G.F.; Liu, T.H. Cultivation of fruiting bodies using hybrid diploids of Armillaria ostoyae. Forest Pest Dis. 2003, 22, 13–16. (In Chinese) [Google Scholar]
- Ford, K.L.; Baumgartner, K.; Henricot, B.; Bailey, A.M.; Foster, G.D. A reliable in vitro fruiting system for Armillaria mellea for evaluation of Agrobacterium tumefaciens transformation vectors. Fungal Biol. 2015, 119, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Fungal species concepts in the genomics era. Genome 2020, 63, 459–468. [Google Scholar] [CrossRef]
- Matute, D.R.; Sepúlveda, V. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef]
- Wolfe, J.A. Some Aspects of Plant Geography of the Northern Hemisphere During the Late Cretaceous and Tertiary. Ann. Mo. Bot. Gard. 1975, 62, 264. [Google Scholar] [CrossRef]
- Ramstein, G.; Fluteau, F.; Besse, J.; Joussaume, S. Effect of orogeny, plate motion and land–sea distribution on Eurasian climate change over the past 30 million years. Nat. Cell Biol. 1997, 386, 788–795. [Google Scholar] [CrossRef]
- Chillali, M.; Wipf, D.; Guillaumin, J.; Mohammed, C.; Botton, B. Delineation of the European Armillaria species based on the sequences of the internal transcribed spacer (ITS) of ribosomal DNA. New Phytol. 1998, 138, 553–561. [Google Scholar] [CrossRef]
- Baumgartner, K.; Travadon, R.; Bruhn, J.; Bergemann, S.E. Contrasting patterns of genetic diversity and population structure of Armillaria mellea sensu stricto in the eastern and western United States. Phytopathology 2010, 100, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Guillaumin, J.J.; Mohammed, C.; Abomo, N.S. Vegetative incompatibility and sexual systems of Armillaria isolates from tropical Africa. In Proceedings of the Eighth International Conference on Root and Butt Rots, Haikko, Finland, 9–16 August 1993; pp. 349–354. [Google Scholar]
- Elías-Román, R.D.; Guzmán-Plazola, R.A.; Klopfenstein, N.B.; Alvarado-Rosales, D.; Calderón-Zavala, G.; Mora-Aguilera, J.A.; Kim, M.-S.; García-Espinosa, R. Incidence and phylogenetic analyses of Armillaria spp. associated with root disease in peach orchards in the State of Mexico, Mexico. For. Pathol. 2013, 43, 390–401. [Google Scholar] [CrossRef]
- Owaid, M.N.; Muslat, M.M.; Tan, W.C. First collection and identification of wild mushrooms in western Iraq. J. Adv. Res. Biol. 2014, 5, 29–34. [Google Scholar]
- Koch, R.A.; Herr, J.R. Global distribution and richness of Armillaria and related species inferred from public databases and amplicon sequencing datasets. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Gavrilets, S. Perspective: Models of speciation: What have we learned in 40 years? Evolution 2003, 57, 2197–2215. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D. Evidence for Ecological Speciation and Its Alternative. Science 2009, 323, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Sipos, G.; Prasanna, A.N.; Walter, M.C.; O’Connor, E.; Bálint, B.; Krizsán, K.; Kiss, B.; Hess, J.; Varga, T.; Slot, J.; et al. Genome expansion and lineage-specific genetic innovations in the forest path-ogenic fungi Armillaria. Nat. Ecol. Evol. 2017, 1, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, E.; Ota, Y.; Hattori, T.; Kikuchi, T. 2010. Sequence based identification of Japanese Armillaria species using elonga-tion factor-1 alpha gene. Mycologia 2010, 102, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.B.; Powell, C.M.; An, Z.S.; Zhou, J.; Dong, G.R. Pliocene uplift of the northern Tibetan plateau. Geology 2000, 28, 715. [Google Scholar] [CrossRef]
- Huntington, K.W.; Blythe, A.E.; Hodges, K.V. Climate change and Late Pliocene acceleration of erosion in the Himalaya. Earth Planet. Sci. Lett. 2006, 252, 107–118. [Google Scholar] [CrossRef]
- Song, C.; Hu, S.; Han, W.; Zhang, T.; Fang, X.; Gao, J.; Wu, F. Middle Miocene to earliest Pliocene sedimentological and geo-chemical records of climate change in the western Qaidam Basin on the NE Tibetan Plateau. Palaeogeogr. Palaeocl. 2014, 395, 67–76. [Google Scholar] [CrossRef]
- Bing, J.; Han, P.J.; Liu, W.Q.; Wang, Q.M.; Bai, F.Y. Evidence for a Far East Asian origin of lager beer yeast. Curr. Biol. 2014, 24, R380–R381. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zheng, Q.J.; Qian, Z.Q.; Yang, J.; Zhang, Y.P.; Li, Z.H.; Zhao, G.F. Genetic structure and evolutionary history of three alpine sclerophyllous Oaks in East Himalaya-Hengduan Mountains and adjacent regions. Front. Plant Sci. 2016, 7, 1688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, S. Extinction vs. Rapid radiation: The juxtaposed evolutionary histories of Coelotine spiders support the Eocene-Oligocene Orogenesis of the Tibetan Plateau. Syst. Biol. 2017, 66, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gladenkov, A.Y.; Oleinik, A.E.; Marincovich, L.; Barinov, K.B. A refined age for the earliest opening of Bering Strait. Palaeogeogr. Palaeoclim. Palaeoecol. 2002, 183, 321–328. [Google Scholar] [CrossRef]
- Leal, I.; Bergeron, M.-J.; Feau, N.; Tsui, C.; Foord, B.; Pellow, K.; Hamelin, R.; Sturrock, R.N. Cryptic speciation in Western North America and Eastern Eurasia of the pathogens responsible for Laminated root rot. Phytopathology 2019, 109, 456–468. [Google Scholar] [CrossRef]
- Li, D.W. Release and dispersal of basidiospores from Amanita muscaria var. alba and their infiltration into a residence. Mycol. Res. 2005, 109, 1235–1242. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).