1. Introduction
The wound-healing process is a complex sequence of cellular and molecular processes that consist of inflammation, new tissue formation, and tissue remodeling [
1,
2]. In brief, there are four stages of the wound-healing process. Hemostasis is the first immediate step that occurs at the wound site and requires platelets to repair damaged blood vessels through the process of blood clotting. The second step involves the inflammation utilized by neutrophils and macrophages in response to tissue injury via cytokine release and phagocytosis [
3]. On approximately the 4th day after wounding, the proliferation phase begins. This process is characterized by the involvement of the cells embedded in the skin layers including specialized fibroblasts, which secrete a collagen framework onto the dermal skin layer in order to facilitate dermal regeneration. In addition, at the epidermal layer, keratinocytes are the cells responsible for epithelialization of the stratum corneum at skin layer resulting in the epidermal regeneration and the wound contraction. Lastly, the remodeling step involves the remodeling and realignment of the collagen tissue. This step is essential in establishing the greater tensile strength of the skin [
4,
5].
Regarding the cellular mechanism of the wound-healing process, tremendous progress has been made in recent years in identifying the critical events responsible for wound healing. Acute and chronic wounds involve a variety of interactions with components of the dermis and epidermal regeneration. Fibroblast cells play an important role in facilitating the proliferative stage via the production of various enzymes that degrade the fibrin clot, then replace it with extracellular matrix (ECM) components (such as collagen and hyaluronic acid) and infiltrate the margins of the wound [
4]. Not only fibroblasts that are essential for the wound-healing process, but keratinocytes are indeed involved in critical steps, such as the epidermal regeneration and the closure of the wound. These steps contribute to the restoration of skin functions and mechanistically required the proliferation, migration, and differentiation of keratinocytes at the margins of the wound [
1,
6]. Despite the fact that the regular wound-healing process can simultaneously occur, many of intrinsic and extrinsic factors can interfere with the wound-healing process that can then result in an ultimate delay of wound repair. These factors include infection, necrotic tissue, vascular supply, co-existing physical factors (nutritional status), and disease states (diabetes, cancer, and arthritis) [
7]. Therefore, an attempt to identify any remedies that could effectively accelerate skin wound healing would be of significant interest.
In recent years, several researchers have investigated various kinds of natural products with regard to their wound-healing properties. These studies were conducted by targeting relevant potential wound repair mechanisms such as the activation of epithelial cells- or fibroblasts proliferation and migration, the inhibition of reactive oxygen species (ROS) production, and the modulation of inflammatory mediators [
8]. Interestingly, the wound-healing properties of plant and fungi extracts have been reported by several authors. For instance, in plants, the aqueous leaf extract of
Ficus exasperata (Moraceae) demonstrated wound-healing activity in rats with chronic diseases [
9]. The perennial plant,
Cissus quadrangularis, displayed wound-healing properties in mice with the skin structure of the wound area being completely returned to physiological state [
10]. Moreover, in mushrooms,
Antrodia camphorata rich in total polyphenols and flavonoids, significantly promoted wound healing in vitro by promoting fibroblast cell proliferation and increasing collagen synthesis in injured tissues in vivo with less inflammatory cell accumulation [
11]. Moreover, the topical application of 10%
w/
w polysaccharide-rich extract derived from the Lingzhi mushroom,
Ganoderma lucidumm, could enhance wound healing in STZ-induced diabetic rats [
12]. Despite the increasing number of studies on the wound-healing activity of mushrooms, the wound-healing mechanism has not yet been fully identified.
Auricularia auricula-judae is a species of edible Auriculariales mushroom. It belongs to the family Auriculariaceae and can be identified by its ear-like shape and brown color. It is commonly referred to as Jew’s ear or (black) wood ear mushroom. It is widely distributed throughout Europe, East-, South-, and Southeast Asia. Besides possessing proteins and minerals,
Auricularia auricula largely contains carbohydrates. The recent reports have shown that
A. auricula extract exhibited antioxidant activity and promoted procollagen biosynthesis [
13], antitumor activities [
14], and anticoagulant activity [
15]. However, the medicinal properties of
A. auricula-judae as a wound-healing agent has not yet been fully elucidated, as only a few studies have been conducted so far. Therefore, this study aimed to investigate the wound-healing potential and underlying mechanism of the polysaccharide-rich extract of
A. auricula-judae in both in vitro and in vivo experiments.
2. Materials and Methods
2.1. Reagents and Chemicals
Dulbecco’s modified Eagle medium (DMEM), trypsin, and penicillin-streptomycin were supplied by Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS), RIPA buffer, protease inhibitors, and Coomassie Plus™ Protein Assay Reagent were obtained from Thermo Scientific Company (Waltham, MA, USA). Nitrocellulose membrane and Enhanced chemiluminescent (ECL) reagent were supplied by GE Healthcare (Little Chalfont, UK). Antibody specific to E-cadherin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Furthermore, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye, ABTS [2,2′-azino-bis (ethylbenzthiazoline-6-sulfonic acid)], 2′,7′-dichlorofluorescin diacetate (DCF-DA), fibronectin, and the antibody to β-actin were purchased from Sigma-Aldrich (St. Louis, MO, USA (Darmstadt, Germany). SephacrylTM S-400 high resolution was purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). Dextran blue derived from Leuconostoc spp. was purchased from Fluka Biochemika (Buchs, Switzerland). Gel filtration standard components (Thyroglobulin, γ-globulin, Ovalbulmin, Myoglobulin, and Vitamin B12) were purchased from Bio-Rad Laboratories, Inc. (Hercules, CA, USA).
2.2. Cell Culture
HaCat cell line (human keratinocytes) was purchased from ATCC (Virginia, United States). Primary human skin fibroblasts were aseptically isolated from an abdominal scar after a surgical procedure involving a Caesarean section that had been performed at the surgical ward of Chiang Mai Maharaj Hospital, Chiang Mai University, Thailand (Study code: BIO-2558-03549 approved of by Medical Research Ethics Committee, Chiang Mai University). The methods of primary fibroblast preparation and culturing were employed following the previously described protocol [
16]. All cell types were cultured in DMEM with 10% FBS, and supplemented with 100 U/mL penicillin, and 100 µg/mL streptomycin. The cells were maintained in a 5% CO
2 humidified incubator at 37 °C.
2.3. Animals
Six- to eight-week-old BALB/c mice (male, weight = 28–30 g) were purchased from Nomura Siam International, Bangkok, Thailand. Mice were acclimatized to the animal housing laboratory for at least 7 days. The animals were housed in specific pathogen-free facilities and handled according to the institutionally recommended guidelines. All animals were maintained under a constant 12 h/12 h light-dark cycle with humidity levels maintained at 60 ± 10% at 25 ± 1 °C. All mice were supplied with free access to water and food.
2.4. A. auricula-judae Extraction
Fresh
A. auricula-judae was harvested by Siri Mushroom Organic Farm in Thung Hua Chang District, Lumphun Province, Thailand. The voucher specimen number of
A. auricula-judae (No. 023241) was certified at the herbarium of the Flora of Thailand, Chiang Mai University, Thailand. The
A. auricula-judae extraction was prepared by following the previously reported procedure with some modifications [
17]. Briefly, the fruiting bodies of
A. auricula-judae were washed and then oven-dried at 50 °C and finally ground into a fine powder. A 100 g sample of
A. auricula-judae dried powder was suspended in 1 L of methanol for 2 h at 76 °C. Thereafter, the suspension was filtered through Whatman No.2 filter paper to remove the methanol-soluble materials. The residue was then resuspended in 1 L of distilled water for 2 h at 97 °C. The clear supernatant was collected after centrifugation at 7800×
g for 15 min at 4 °C. This extraction step was repeated three times and the obtained supernatants were pooled and concentrated in a rotary evaporator. The concentrated supernatant was filtrated to remove any water-insoluble materials. The crude polysaccharide was obtained by precipitation in ethanol for 24 h, at 4 °C. The resulting precipitate was kept after centrifugation at 7800×
g for 15 min at 4 °C. After that, the crude polysaccharide was dried at room temperature, dissolved in distilled water, and dialyzed against running water for 24 h. The non-dialyzed portion was centrifuged to remove any insoluble materials, then lyophilized and named as an
A. auricula-judae polysaccharide-rich extract (AAP).
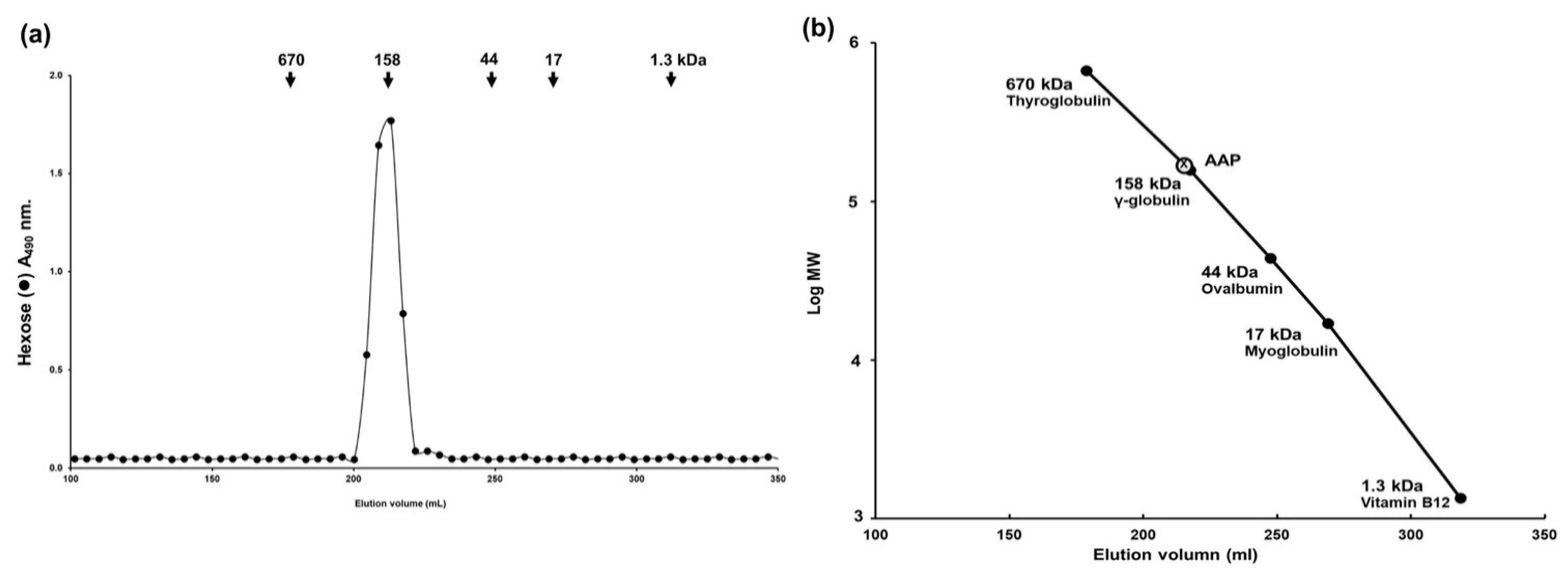
2.5. Molecular Weight of Polysaccharide Derived from A. auricula-judae Determination by Sephacryl S-400 Gel Filtration
To determine the molecular weight of AAP, the extract (2.5 mg) was dissolved in 0.5 mL of 0.2 M ammonium bicarbonate buffer (pH 7.0) and applied onto a Sephacryl S-400 HR column (1.7 × 130 cm) equilibrated with 0.2 M ammonium bicarbonate buffer. The column was eluted with the same buffer at a flow rate of 25 mL/h and the aliquots were collected as 5 mL per tube. These fractions were assayed for hexose sugars and for standard proteins, as will be described in the next section (wavelength absorbance of 490 nm and 280 nm, respectively) [
17]. The column was calibrated using Blue dextran (average MW = 2000 kDa, for Vo), Phenol red (MW = 354 Da, for Vt), and standard proteins with different molecular masses (Thyroglobulin MW = 670 kDa; γ-globulin MW = 158 kDa; Ovalbumin MW = 44 kDa; Myoglobin MW = 17 kDa; and Vitamin B12 MW = 1.3 kDa). The molecular weight of AAP was calculated using the log molecular weight (logMW) curve of standard proteins.
2.6. Total Carbohydrate Assay
The total carbohydrate content in AAP was determined using a Total Carbohydrate Assay Kit (Sigma-Aldrich, USA) according to the manufacturer’s protocol. Briefly, various concentrations of AAP (30 µL) and sulfuric acid (150 µL) were added to each of the tubes. The solutions were then mixed by pipetting and the reaction was incubated for 15 min at 90 °C in the dark. A developer was added to each well and they were mixed using a shaker for 5 min at room temperature. Subsequently, the contents were mixed for 1 min before being measured at an absorbance of 490 nm.
2.7. Quantification of the Constituent Monosaccharides A. auricula-judae Using GC–MS
An AAP sample was prepared by following the previously described protocol for GC-MS analysis [
18]. Dried samples were methanolized with 3 N HCl in methanol for 2 h at 121 °C followed by re-N-acetylation and tri-methylsilylation. An analysis of the constituent monosaccharides was performed by following the previously reported protocol [
19]. Briefly, helium was used as the carrier gas at a flow rate of 1 mL/min. The temperature program was set to increase from an initial 120 to 200 °C, at 8 °C/min and held for 10 min before being heated at 6 °C/min to 230 °C. It was then held for a further 20 min. Peaks were identified and estimated using myoinositol as the internal standard. The quantity of fractions was determined from the peak area using response factors. The inlet temperature was kept constant at 210 °C and the MS transfer line was set at 270 °C. Mass spectrophotometer (MS) acquisition parameters included scanning from
m/z 50 to 550 in the electron impact (EI) mode for routine analysis.
2.8. Antioxidant Activity by ABTS Assay
The antioxidant activity of the AAP was determined using ABTS assay. The ABTS radical cation was prepared by mixing 7 mM ABTS stock solution with 2.45 mM potassium persulfate (K
2S
2O
8) (1/1,
v/
v). The mixture was incubated in the dark for 12–16 h until the reaction was completed. The assay was conducted on 990 µL of ABTS solution and 10 µL of the AAP (0–400 μg/mL), and vitamin E was used as a positive control in this assay. After 6 min of incubation, the absorbance was recorded immediately at 734 nm using a spectrophotometer. The percent inhibition of ABTS activity was calculated using the following equation:
2.9. Intracellular ROS Determination
Intracellular ROS in cellulo upon UVB irradiation was determined using DCF-DA as has been previously described [
20]. Primary fibroblasts (8.0 × 10
3 cells/well) were seeded in a 96-well plate for 24 h. After that, cells were exposed to UVB at the intensity of 15 mJ/cm
2 using the ultraviolet cross linker (CL-1000, UPV Inc., Upland, CA, USA). Fibroblast cells were then treated with or without increasing the concentrations of AAP (0–100 µg/mL) or 25 µg/mL of vitamin C (positive control) dissolved in culture medium for the duration of 24 h. Fibroblasts were then washed with PBS, 10 µM of DCF-DA was then added and the cells were incubated for a further 30 min. The fluorescent intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 525 nm. Intracellular ROS was calculated and compared to the control of the UVB-irradiated fibroblast cells.
2.10. Cell Proliferation Assay
The effects of AAP on cell proliferation were determined by MTT and trypan blue staining assays. For the MTT assay, the cells (8 × 103 cells/well) were treated with increasing concentrations of AAP (0–25 μg/mL) in culture medium or culture medium alone (vehicle control) for 24 and 48 h. Following the AAP treatment, the cells were incubated with 10 μL of 0.5 mg/mL MTT in PBS for 4 h. The culture supernatant was then removed, and the culture was re-suspended with 200 μL of DMSO to dissolve the MTT formazan crystals. The absorbance was measured at 540 and 630 nm using a UV-visible spectrophotometer. The assay was performed in triplicate at each concentration.
With regard to the trypan blue cell staining method, the cells (1.5 × 105 cells/well) were seeded in a 6-well plate and incubated for 24 h. Further, the cells were treated with different concentrations of AAP (0–25 µg/mL) in culture medium or culture medium alone (vehicle control) and incubated for 24 and 48 h. Finally, cells were trypsinized and suspended in the culture medium. The cells were stained with 0.4% trypan blue dye at the ratio of 1:1. Cell proliferation for each concentration was determined using trypan blue dye and the resulting values were compared with the control.
2.11. Scratch Assay
Wound-healing activity was examined using the modified scratch assay as has been previously described [
21]. The primary fibroblast (2 × 10
5 cells/well) or HaCat cells (2 × 10
5 cells/well) were seeded into each well of a 6-well plate and incubated with completed DMEM medium at 37 °C and 5% CO
2 for 24 h. After incubation, the monolayer confluent cells were scrapped horizontally with a sterile yellow pipette tip and washed with PBS. The cells were incubated with various concentrations of AAP (0–25 µg/mL) by diluting with 0.5% FBS-DMEM or 0.5% FBS-DMEM without AAP (vehicle control). The migration rates were observed, photographed under phase-contrast microscopy, and analyzed using IMAGE J 1.410. The percentage of the closed area was measured and then compared with the control.
2.12. Transwell Migration and Invasion Assays
The effect of AAP on human fibroblast and keratinocyte cell migration, and invasion, was determined by transwell assay as has been previously described [
22,
23]. Polyvinylpyrrolidone-free polycarbonate filters (8 μM pore size) (BD Biosciences, Franklin Lakes, NJ, USA) were coated with 10 μg/mL fibronectin (migration assay) along with 15 μg of matrigel per filter (invasion assay). The primary fibroblasts (1 × 10
5 cells/well) and HaCat cells (2 × 10
5 cells/well) were treated with various concentrations of AAP (0–25 µg/mL) in 0.5% FBS-DMEM or 0.5% FBS-DMEM without AAP (vehicle control) and seeded into the upper chamber. The 10% FBS-DMEM was added into the lower chamber as a chemoattractant. The cells were incubated for the next 48 h for both invasion and migration assays. The invading or migrating cells were fixed with 95% ethanol for 5 min. They were then stained with 0.5% crystal violet in 20% methanol for 30 min. The invading cells were photographed under phase-contrast microscopy (Nikon Eclipse TS100, Nikon Instrument Inc., Melville, NY, USA). The percentages of the areas occupied with cells were then determined with IMAGE J 1.410. The percentage of cell invasion or migration was measured and compared with control.
2.13. Collagen Synthesis Assay
The primary fibroblast cells (1 × 105 cells/well) were seeded into 24-well plates for 24 h and incubated at 37 °C in an atmosphere of 5% CO2. After incubation, the cells were pre-treated with the 0.5%-FBS-DMEM medium for 24 h. The medium was then removed, and the cells were incubated with or without AAP (0–25 µg/mL) or vitamin C; 25 µg/mL (a positive control), in 0.5%-FBD-DMEM (vehicle control) for 48 h. After that, the cultured medium was kept and the collagen was determined using Sirius Red Collagen Staining Kit (Chondrex Inc., Redmond, WA, USA) according to the manufacturer’s protocol. Briefly, the proteins in the culture supernatant were precipitated using 25% tricholoacetic acid (TCA) and washed with 5% TCA. Proteins were dissolved in 0.05 M acetic acid. The collagen was then stained with Sirius red for 20 min at RT and washed with washing buffer. The collagen pellets were dissolved in an extraction buffer and were then measured at 540 nm using a UV-visible spectrophotometer. The collagen in each sample was calculated from the collagen standard curve and interpreted as the % of control.
2.14. Western Blot Analysis
The primary fibroblasts (2.0 × 105 cells/well) and the HaCat keratinocyte cells (4.5 × 105 cells/well) were treated with increasing concentrations of AAP for 24 and 48 h. After incubation, the cells were collected and lysed using RIPA buffer. Protein concentration levels were determined using the Bradford method. The whole-cell lysates were then subjected to 10% SDS-PAGE. The proteins were transferred onto nitrocellulose membranes and the membranes were blocked with 5% non-fat dried milk protein in 0.5% TBS-tween. After that, the membranes were further incubated overnight with the desired primary antibody at 4 °C followed by incubation with horseradish peroxidase-conjugated secondary antibody. Bound antibodies were detected using the chemiluminescent detection system and then exposed to the X-ray film (GE Healthcare Ltd., Little Chalfont, UK). The bands were normalized to β-actin as a loading control. Band density levels were analyzed using IMAGE J 1.410 (National Institutes of Health, Rockville, MD, USA).
2.15. In Vivo Wound-Healing Activity by Mice Skin Wound-Healing Model
An assessment of the wound lesion area was done to investigate the wound-healing activity of AAP. This was examined in the mice skin wound-healing model using the modified protocol that has been previously described [
24]. In brief, BABL/c mice were subjected to full skin thickness excision. Mice were anesthetized before circular wounds of 5 mm in diameter were created on the shaved dorsum of the mice using a disposable biopsy punch (Kai medical, Japan). Twenty-one wounded mice were randomly divided into three groups. Every group was treated with either sterilized 0.9% normal saline (vehicle control) or an appropriate dose of AAP (1.0%
w/
v, or 2.5%
w/
v). To monitor the wound closure, photographs of the mice were taken every three days and the wound areas were measured using a digital vernier calipers (BEC, Taiwan). The wound area of each mouse was calculated using the following formula:
Experimental mice were sacrificed on day 12 of the experiment. Wounded tissues were then harvested and fixed for histological analysis with 10% formalin buffer solution and embedded in paraffin. A section of 4 μm thickness was prepared and stained with either hematoxylin and eosin (H&E) or Masson’s trichrome. The stained sections were observed under a digital whole-slide scanner (Pannoramic MIDI II machine, 3DHISTECH Ltd., Budapest, Hungary) and photographed (10×).
2.16. Statistical Analysis
All data are presented as mean ± standard deviation (S.D.) or standard error (S.E.) values. Statistical analysis was conducted with GraphPad Prism 6.0 using independent t-test or one-way ANOVA with Dunnett’s test. Statistical significance was determined at p < 0.05, p < 0.01, and p < 0.001.
4. Discussion
The results of our study indicate that AAP accelerates the wound-healing process through the promotion of fibroblast and keratinocyte proliferation, migration, and invasion, and the enhancement of collagen synthesis that occurs during the proliferation step of the wound repair process. As was consistent with our study, other medicinal mushroom and plant-based extracts have been verified for wound-healing properties with those targeting various mechanisms, including epithelial and dermal cells stimulation, reduction of reactive oxygen species (ROS), and modulation of inflammatory intermediates [
25,
26]. The efficiency of the wound-healing process is largely dependent upon the balance of proinflammatory and pro-regenerative signals that are mediated by cytokines [
27,
28]. These statements support our findings as AAP was found to display strong antioxidant properties that may have beneficial effects on balancing the pro- and anti- inflammatory mediators of the local wound environment. In previous studies,
A. auricula mushroom extracts exhibited a variety of pharmacological properties such as antioxidant, blood lipid-lowering, anti-inflammation, antitumor, and anti-radiation activities [
13,
19,
29,
30,
31]; however, there is no direct evidence for their wound-healing effects. The levels of ROS in tissue injuries are considered to be associated with the wound-healing processes. Low levels of ROS are essential for the stimulation of wound healing. However, high levels of ROS can impair ROS detoxification resulting in cellular damage and aggravation of the wound [
32]. Moreover, intracellular ROS generation is one of the important factors that promote prolonged inflammation occurring in the surrounding wound margins [
33]. Previously, experimental and clinical studies have indicated that those antioxidant and anti-inflammatory properties are considered beneficial elements at the initial stage of wound healing [
32,
34]. In this study, the antioxidant properties of AAP were confirmed by both in vitro ABTS assay and in cellulo DCF-DA fluorescent assay. Moreover, AAP exhibited stronger antioxidant activity when compared with the other plant-based polysaccharide-rich extracts including
Phyllophorus proteus (EC
50 > 1 mg/mL) [
31] and
Ganoderma lucidum (EC
50 > 1 mg/mL) [
35]. Importantly, AAP has been proven to possess antioxidant functions that assist in the reduction of skin inflammation.
Regarding the proliferation phase of the wound-healing process, new tissue formation is characterized by human fibroblast and keratinocyte proliferation, as well as their migration and invasion [
4]. In contrast, impaired keratinocyte migration results in poor wound healing and the persistence of chronic wounds [
23]. Our results demonstrated that human fibroblast and keratinocyte cells proliferation, migration, and invasion were induced by AAP in a dose-dependent manner. These results indicate wound-healing potential of AAP. Moreover, E-cadherin is the epithelial marker, which mediated cell–cell adhesion of epithelial cells. Previous studies reported that, the downregulation of E-cadherin promoted wound healing via increased epithelial cells invasion/migration [
23,
36]. Our results showed that AAP reduced the E-cadherin expressions in keratinocytes. Therefore, the downregulation of E-cadherin might be one of the wound-healing mechanisms of AAP. Apart from keratinocytes, during the proliferation state, fibroblasts also regulated the synthesis of the new ECM by secretion of collagen and other ECM proteins [
2,
4]. Our result showed that AAP could significantly stimulate collagen synthesis from human fibroblasts. Thus, the stimulation of collagen synthesis might be considered as another important wound-healing mechanism of AAP.
Based on the results of Sephacryl-S400 gel filtration and GC-MS studies, we can conclude that AAP is a homogeneous polysaccharide with a molecular weight of 158 kDa. The outcomes of our study strongly suggest that the wound-healing properties of AAP might be related to the rich content of polysaccharides in the extract. Our findings are in agreement with those of another study which reported on the molecular weight of
A. auricula mushrooms [
17]. Furthermore, the monosaccharides found in AAP (mannose, glucose, and galactose) were previously reported as either active compounds or a type of sugar backbone in the wound-healing agents based on the mushroom’s origins [
37,
38]. In terms of wound-healing properties, our data coincided with that of other reports which had used polysaccharide-rich extracts of either plants (
Bletilla striata and
Astragalus membranaceus) [
39,
40], seaweed (
Gracilaria lemaneiformis) [
24] or mushrooms (
Ganoderma lucidum, Agaricus blazei and
Phellinus gilvus) [
41,
42] to enhance keratinocyte and fibroblast cells proliferation or to accelerate the wound healing with other appropriate mechanisms [
25]. Herein, this is the first report to demonstrate that polysaccharide-rich extract can stimulate the proliferation, migration, and invasion of skin keratinocytes and fibroblasts.
Our in vivo findings support the wound-healing potential of AAP as it could accelerate wound closure and improve the appearance of wound architecture without any cytotoxic effect in the mice skin wound-healing model. According to previous reports, skin that was either damaged or wounded was healed by the migration of the three main types of skin cells: keratinocytes, fibroblasts, and endothelial cells [
43,
44]. Without the migration of these cells, the main processes of wound healing cannot occur. This can lead to an unfinished wound-healing process and result in the delay of wound closure and the persistence of inflammation, which can then cause the formation of a chronic wound. Findings of previous in vivo studies on the wound-healing effects of medicinal mushrooms are in agreement with our results. Specifically, the polysaccharides obtained from
Gracilaria lemaneiformis could induce wound healing in KM mice by accelerating the wound contraction rate, improving the epithelial layer thickness and collagen deposition, and decreasing inflammation [
24]. Our data are the first report to display the in vivo wound-healing properties of polysaccharides obtained from the medicinal mushroom
A. auricula-judae. Overall, our data reveal the benefits associated with the use of AAP as a potential candidate for wound-healing-accelerating agents due to its remarkable wound-healing properties identified in both in vitro and in vivo experiments.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





