
Alternative Transcription Start Site Usage and Functional Implications in Pathogenic Fungi


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
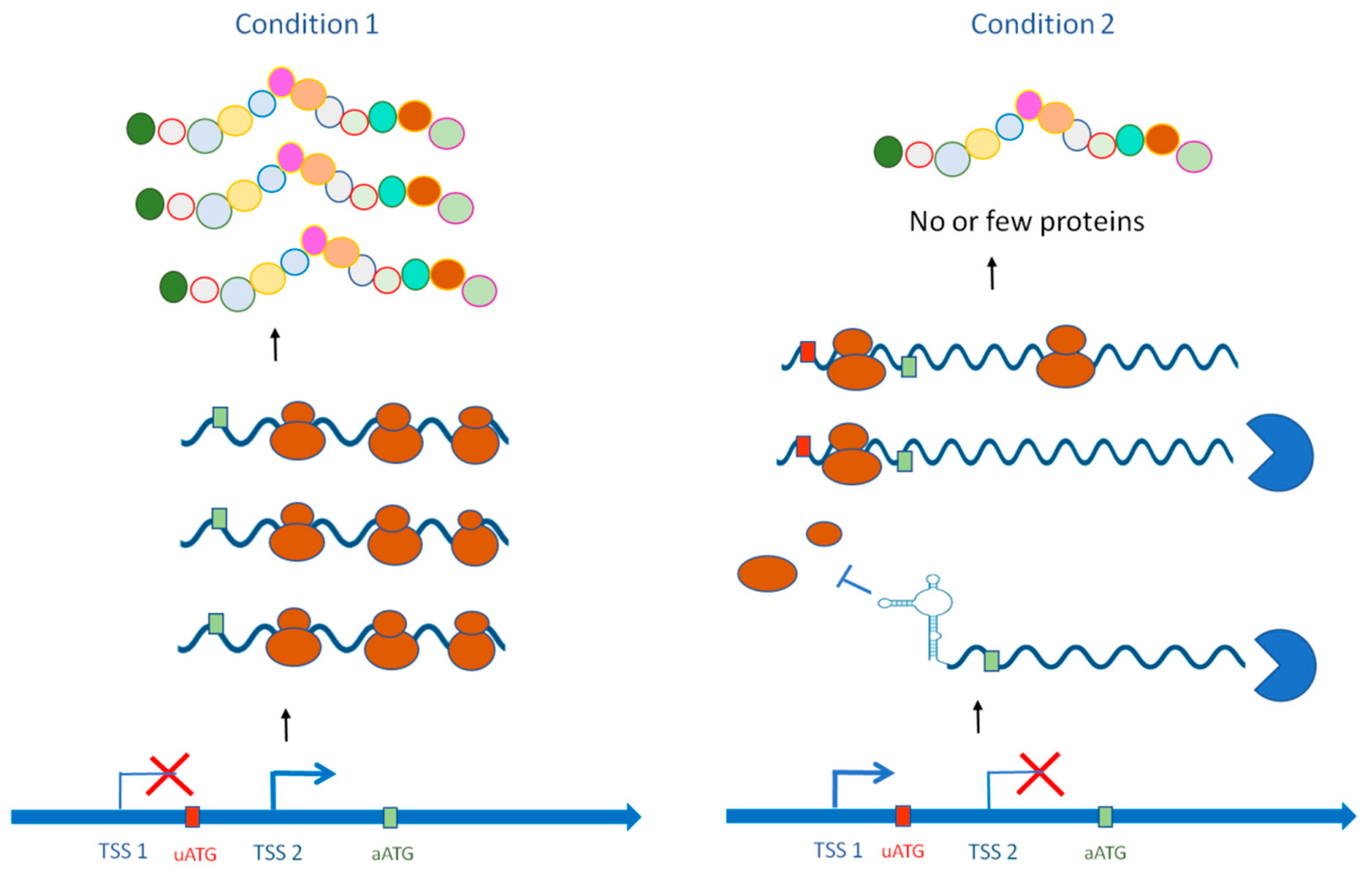
Abstract
Share and Cite
Dang, T.T.V.; Colin, J.; Janbon, G. Alternative Transcription Start Site Usage and Functional Implications in Pathogenic Fungi. J. Fungi 2022, 8, 1044. https://doi.org/10.3390/jof8101044
Dang TTV, Colin J, Janbon G. Alternative Transcription Start Site Usage and Functional Implications in Pathogenic Fungi. Journal of Fungi. 2022; 8(10):1044. https://doi.org/10.3390/jof8101044
Chicago/Turabian StyleDang, Thi Tuong Vi, Jessie Colin, and Guilhem Janbon. 2022. "Alternative Transcription Start Site Usage and Functional Implications in Pathogenic Fungi" Journal of Fungi 8, no. 10: 1044. https://doi.org/10.3390/jof8101044
APA StyleDang, T. T. V., Colin, J., & Janbon, G. (2022). Alternative Transcription Start Site Usage and Functional Implications in Pathogenic Fungi. Journal of Fungi, 8(10), 1044. https://doi.org/10.3390/jof8101044