
Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years



{kind=link}
Abstract
1. Introduction
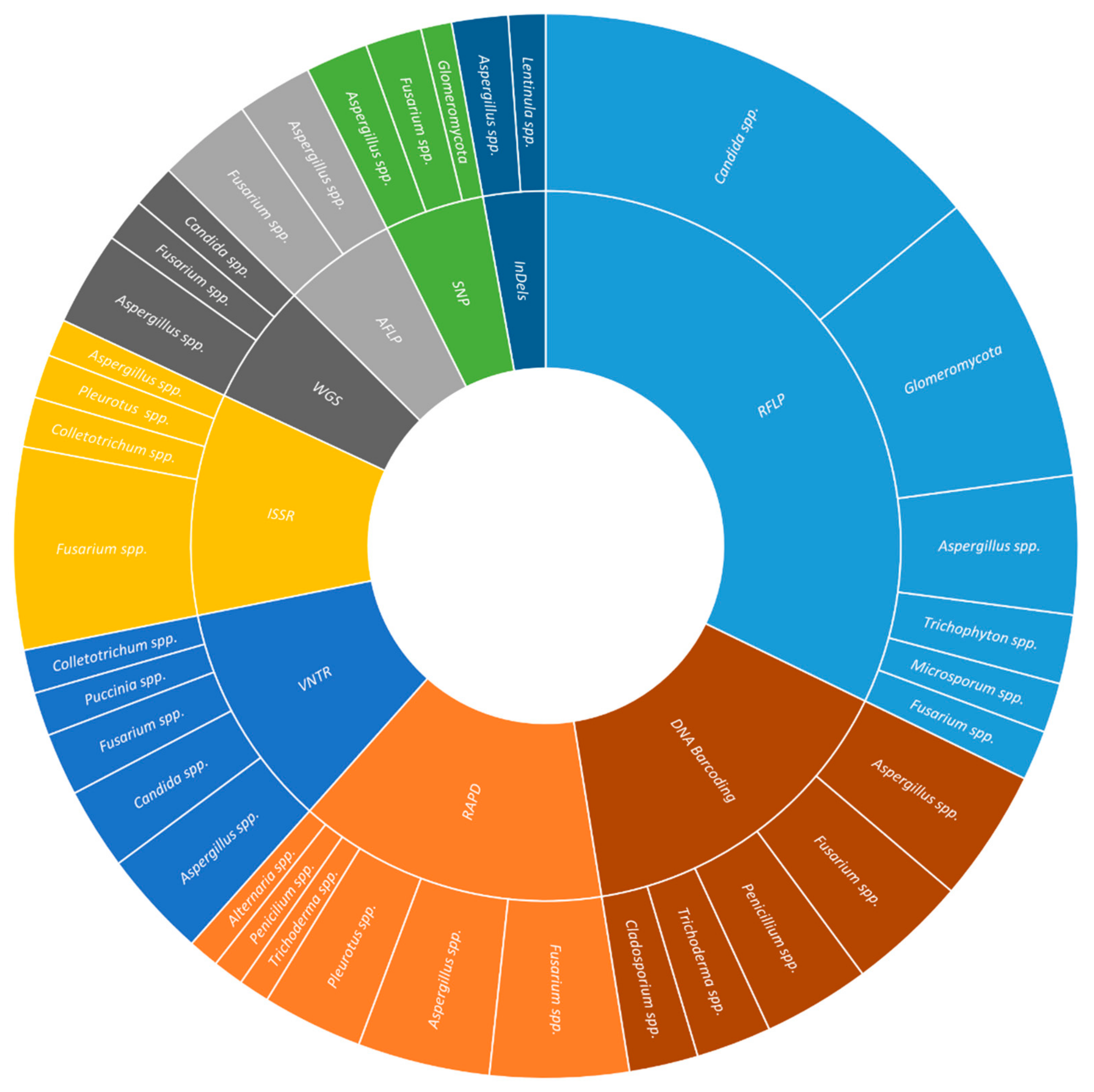
2. DNA-Based Molecular Markers
2.1. Restriction Fragment Length Polymorphism (RFLP)
2.2. Random Amplification of Polymorphic DNA (RAPD)
2.3. Amplified Fragment Length Polymorphism (AFLP)
2.4. Inter-Simple Sequence Repeats (ISSR)
2.5. Variable Number of Tandem Repeats (VNTR)
2.6. Single-Nucleotide Polymorphisms (SNP)
2.7. Small Insertions or Deletions (InDels)
2.8. DNA Barcoding
2.9. Massive Parallel Sequencing (MPS)
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmit, J.P.; Mueller, G.M. An estimate of the lower limit of global fungal diversity. Biodivers. Conserv. 2007, 16, 99–111. [Google Scholar] [CrossRef]
- Blackwell, M. The Fungi: 1, 2, 3… 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10. [Google Scholar] [CrossRef]
- Vu, D.; Groenewald, M.; de Vries, M.; Gehrmann, T.; Stielow, B.; Eberhardt, U.; Al-Hatmi, A.; Groenewald, J.Z.; Cardinali, G.; Houbraken, J.; et al. Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud. Mycol. 2018, 91, 23–36. [Google Scholar] [CrossRef]
- Adnan, M.; Islam, W.; Gang, L.; Chen, H.Y. Advanced research tools for fungal diversity and its impact on forest ecosystem. Environ. Sci. Pollut. Res. 2022, 29, 45044–45062. [Google Scholar] [CrossRef]
- Barros, J.; Seena, S. Fungi in Freshwaters: Prioritising Aquatic Hyphomycetes in Conservation Goals. Water 2022, 14, 605. [Google Scholar] [CrossRef]
- Gonçalves, M.F.; Esteves, A.C.; Alves, A. Marine Fungi: Opportunities and Challenges. Encyclopedia 2022, 2, 559–577. [Google Scholar] [CrossRef]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Shamim, M.; Kumar, P.; Kumar, R.R.; Kumar, M.; Kumar, R.R.; Singh, K.N. Assessing Fungal Biodiversity Using Molecular Markers. In Molecular Markers in Mycology; Fungal Biology; Singh, B.P., Gupta, V.K., Eds.; Springer: Cham, Switzerland, 2017; pp. 305–333. [Google Scholar] [CrossRef]
- Gautam, A.K.; Verma, R.K.; Avasthi, S.; Bohra, Y.; Devadatha, B.; Niranjan, M.; Suwannarach, N. Current insight into traditional and modern methods in fungal diversity estimates. J. Fungi 2022, 8, 226. [Google Scholar] [CrossRef]
- Tahir, A.; Iqbal, I.; Talib, K.M.; Luhuai, J.; Chen, X.; Akbar, A.; Asghar, A.; Ali, I. Modern Tools for the Identification of Fungi, Including Yeasts. In Extremophilic Fungi; Sahay, S., Ed.; Springer: Singapore, 2022; pp. 33–51. [Google Scholar] [CrossRef]
- Chaudhary, R.; Kumar, G.M. Restriction fragment length polymorphism. In Encyclopedia of Animal Cognition and Behavior; Vonk, J., Shackelford, T.K., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Atoui, A.; El Khoury, A. PCR-RFLP for Aspergillus species. In Mycotoxigenic Fungi. Methods in Molecular Biology; Moretti, A., Susca, A., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1542, pp. 313–320. [Google Scholar] [CrossRef]
- Kennedy, N.; Clipson, N. Fingerprinting the fungal community. Mycologist 2003, 17, 158–164. [Google Scholar] [CrossRef]
- Gryta, A.; Frąc, M. Methodological aspects of multiplex terminal restriction fragment length polymorphism-technique to describe the genetic diversity of soil bacteria, archaea and fungi. Sensors 2020, 20, 3292. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.; McClelland, M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990, 18, 7213–7218. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [PubMed]
- Bardakci, F. Random amplified polymorphic DNA (RAPD) markers. Turk. J. Biol. 2001, 25, 185–196. [Google Scholar]
- Matthes, M.C.; Daly, A.; Edwards, K.J. Amplified fragment length polymorphism (AFLP). In Molecular Tools for Screening Biodiversity; Karp, A., Isaac, P.G., Ingram, D.S., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 183–190. [Google Scholar] [CrossRef]
- Blears, M.; de Grandis, S.; Lee, H.; Trevors, J.T. Amplified fragment length polymorphism (AFLP): A review of the procedure and its applications. J. Ind. Microbiol. Biotech. 1998, 21, 99–114. [Google Scholar] [CrossRef]
- Savelkoul, P.H.M.; Aarts, H.J.M.; de Haas, J.; Dijkshoorn, L.; Duim, B.; Otsen, M.; Rademaker, J.L.; Schouls, W.L.; Lenstra, J.A. Amplified-fragment length polymorphism analysis: The state of an art. J. Clin. Microbiol. 1999, 37, 3083–3091. [Google Scholar] [CrossRef]
- Fry, N.K.; Savelkoul, P.H.; Visca, P. Amplified fragment length polymorphism analysis. In Molecular Epidemiology of Microorganisms. Methods in Molecular Biology; Caugant, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 551, pp. 89–104. [Google Scholar] [CrossRef]
- Zietkiewicz, E.; Rafalski, A.; Labuda, D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 1994, 20, 176–183. [Google Scholar] [CrossRef]
- Hassel, K.; Gunnarsson, U. The use of inter simple sequence repeats (ISSR) in bryophyte population studies. Lindbergia 2003, 28, 152–157. [Google Scholar]
- Jeffreys, A.; Wilson, V.; Thein, S. Hypervariable ‘minisatellite’ regions in human DNA. Nature 1985, 314, 67–73. [Google Scholar] [CrossRef]
- Jeffreys, A.J.; Neumann, R.; Wilson, V. Repeat unit sequence variation in minisatellites: A novel source of DNA polymorphism for studying variation and mutation by single molecule analysis. Cell 1990, 60, 473–485. [Google Scholar] [CrossRef]
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1989, 44, 397. [Google Scholar]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F.; et al. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef]
- Fischer, M.C.; Rellstab, C.; Leuzinger, M.; Roumet, M.; Gugerli, F.; Shimizu, K.K.; Holderegger, R.; Widmer, A. Estimating genomic diversity and population differentiation–an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genom. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Tsykun, T.; Rellstab, C.; Dutech, C.; Sipos, G.; Prospero, S. Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 2017, 119, 371–380. [Google Scholar] [CrossRef]
- Zimmerman, S.J.; Aldridge, C.L.; Oyler-McCance, S.J. An empirical comparison of population genetic analyses using microsatellite and SNP data for a species of conservation concern. BMC Genom. 2020, 21, 382. [Google Scholar] [CrossRef]
- Estoup, A.; Jarne, P.; Cornuet, J.M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002, 11, 1591–1604. [Google Scholar] [CrossRef]
- Lander, E.S. The new genomics: Global views of biology. Science 1996, 274, 536–539. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, Y.-X.; Hewett-Emmett, D.; Boerwinkle, E. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 2003, 312, 207–213. [Google Scholar] [CrossRef]
- Van der Heyden, H.; Dutilleul, P.; Brodeur, L.; Carisse, O. Spatial distribution of single-nucleotide polymorphisms related to fungicide resistance and implications for sampling. Phytopathology 2014, 104, 604–613. [Google Scholar] [CrossRef]
- Kaiser, S.A.; Taylor, S.A.; Chen, N.; Sillett, T.S.; Bondra, E.R.; Webster, M.S. A comparative assessment of SNP and microsatellite markers for assigning parentage in a socially monogamous bird. Mol. Ecol. Resour. 2017, 17, 183–193. [Google Scholar] [CrossRef]
- Dutech, C.; Enjalbert, J.; Fournier, E.; Delmotte, F.; Barres, B.; Carlier, J.; Tharreau, D.; Giraud, T. Challenges of microsatellite isolation in fungi. Fungal Genet. Biol. 2007, 44, 933–949. [Google Scholar] [CrossRef]
- Yang, J.; He, J.; Wang, D.; Shi, E.; Yang, W.; Geng, Q.; Wang, Z. Progress in research and application of InDel markers. Biodivers. Sci. 2016, 24, 237–243. [Google Scholar] [CrossRef]
- Frézal, L.; Leblois, R. Four years of DNA barcoding: Current advances and prospects. Infect. Genet. Evol. 2008, 8, 727–736. [Google Scholar] [CrossRef]
- Lebonah, D.E.; Dileep, A.; Chandrasekhar, K.; Sreevani, S.; Sreedevi, B.; Pramoda Kumari, J. DNA barcoding on bacteria: A review. Adv. Biol. 2014, 2014, 541787. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. DNA barcodes: Methods and protocols. Methods Mol. Biol. 2012, 858, 3–8. [Google Scholar] [CrossRef]
- Valentini, A.; Pompanon, F.; Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol. 2009, 24, 110–117. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Xu, J. Fungal DNA barcoding. Genome 2016, 1, 913–932. [Google Scholar] [CrossRef]
- Mbareche, H.; Dumont-Leblond, N.; Bilodeau, G.J.; Duchaine, C. An overview of bioinformatics tools for DNA meta-barcoding analysis of microbial communities of bioaerosols: Digest for microbiologists. Life 2020, 10, 185. [Google Scholar] [CrossRef]
- Cuomo, C.A. Harnessing Whole Genome Sequencing in Medical Mycology. Curr. Fungal Infect. Rep. 2017, 11, 52–59. [Google Scholar] [CrossRef]
- Oliveira, M.; Amorim, A. Microbial forensics: New breakthroughs and future prospects. Appl. Microbiol. Biotechnol. 2018, 102, 10377–10391. [Google Scholar] [CrossRef]
- Oliveira, M.; Arenas, M.; Amorim, A. New Trends in Microbial Epidemiology: Can An Old Dog Learn New Tricks? Ann. Microbiol. Immunol. 2018, 1, 1004. [Google Scholar]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Fosso, B.; Santamaria, M.; Marzano, M.; Alonso-Alemany, D.; Valiente, G.; Donvito, G.; Monaco, A.; Notarangelo, P.; Pesole, G. BioMaS: A modular pipeline for Bioinformatic analysis of Metagenomic AmpliconS. BMC Bioinform. 2015, 16, 203. [Google Scholar] [CrossRef]
- Banchi, E.; Pallavicini, A.; Muggia, L. Relevance of plant and fungal DNA metabarcoding in aerobiology. Aerobiologia 2020, 36, 9–23. [Google Scholar] [CrossRef]
- Op De Beeck, M.; Lievens, B.; Busschaert, P.; Declerck, S.; Vangronsveld, J.; Colpaert, J.V. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE 2014, 9, e97629. [Google Scholar] [CrossRef]
- Banchi, E.; Stankovic, D.; Fernández-Mendoza, F.; Gionechetti, F.; Pallavicini, A.; Muggia, L. ITS2 metabarcoding analysis complements lichen mycobiome diversity data. Mycol. Prog. 2018, 17, 1049–1066. [Google Scholar] [CrossRef]
- Núñez, A.; de Paz, G.A.; Rastrojo, A.; García, A.M.; Alcamí, A.; Montserrat Gutiérrez-Bustillo, A.; Moreno, D.A. Monitoring of airborne biological particles in outdoor atmosphere. Part 2: Metagenomics applied to urban environments. Int. Microbiol. 2016, 19, 69–80. [Google Scholar] [CrossRef]
- Lear, G.; Dickie, I.; Banks, J.; Boyer, S.; Buckley, H.; Buckley, T.; Cruickshank, R.; Dopheide, A.; Handley, K.M.; Hermans, S.; et al. Methods for the extraction, storage, amplification and sequencing of DNA from environmental samples. N. Z. J. Ecol. 2018, 42, 10–50. [Google Scholar] [CrossRef]
- Bell, K.L.; Burgess, K.S.; Okamoto, K.C.; Aranda, R.; Brosi, B.J. Review and future prospects for DNA barcoding methods in forensic palynology. Forensic Sci. Int. Genet. 2016, 21, 110–116. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Kumar, S.; Carlsen, T.; Mevik, B.H.; Enger, P.; Blaalid, R.; Shalchian-Tabrizi, K.; Kauserud, H. CLOTU: An online pipeline for processing and clustering of 454 amplicon reads into OTUs followed by taxonomic annotation. BMC Bioinform. 2011, 12, 182. [Google Scholar] [CrossRef]
- Gweon, H.S.; Oliver, A.; Taylor, J.; Booth, T.; Gibbs, M.; Read, D.S.; Griffiths, R.I.; Schonrogge, K. PIPITS: An automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol. Evol. 2015, 6, 973–980. [Google Scholar] [CrossRef]
- Gdanetz, K.; Benucci, G.M.N.; Vande Pol, N.; Bonito, G. CONSTAX: A tool for improved taxonomic resolution of environmental fungal ITS sequences. BMC Bioinform. 2017, 18, 1–9. [Google Scholar] [CrossRef]
- Soverini, M.; Turroni, S.; Biagi, E.; Brigidi, P.; Candela, M.; Rampelli, S. HumanMycobiomeScan: A new bioinformatics tool for the characterization of the fungal fraction in metagenomic samples. BMC Genom. 2019, 20, 496. [Google Scholar] [CrossRef]
- El-Kamand, S.; Papanicolaou, A.; Morton, C.O. The use of whole genome and next-generation sequencing in the diagnosis of invasive fungal disease. Curr. Fungal Infect. Rep. 2019, 13, 284–291. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, M.; Azevedo, L. Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years. J. Fungi 2022, 8, 803. https://doi.org/10.3390/jof8080803
Oliveira M, Azevedo L. Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years. Journal of Fungi. 2022; 8(8):803. https://doi.org/10.3390/jof8080803
Chicago/Turabian StyleOliveira, Manuela, and Luísa Azevedo. 2022. "Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years" Journal of Fungi 8, no. 8: 803. https://doi.org/10.3390/jof8080803
APA StyleOliveira, M., & Azevedo, L. (2022). Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years. Journal of Fungi, 8(8), 803. https://doi.org/10.3390/jof8080803