
Transcriptome Profile of Fusarium graminearum Treated by Putrescine


Abstract
:1. Introduction
2. Materials and Methods
2.1. F. graminearum Strain and Growth Conditions
2.2. RNA Extraction, Library Preparation, and Sequencing
2.3. Quality Control and Data Assembly
2.4. Analysis of DEGs and Functional Annotation
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Results
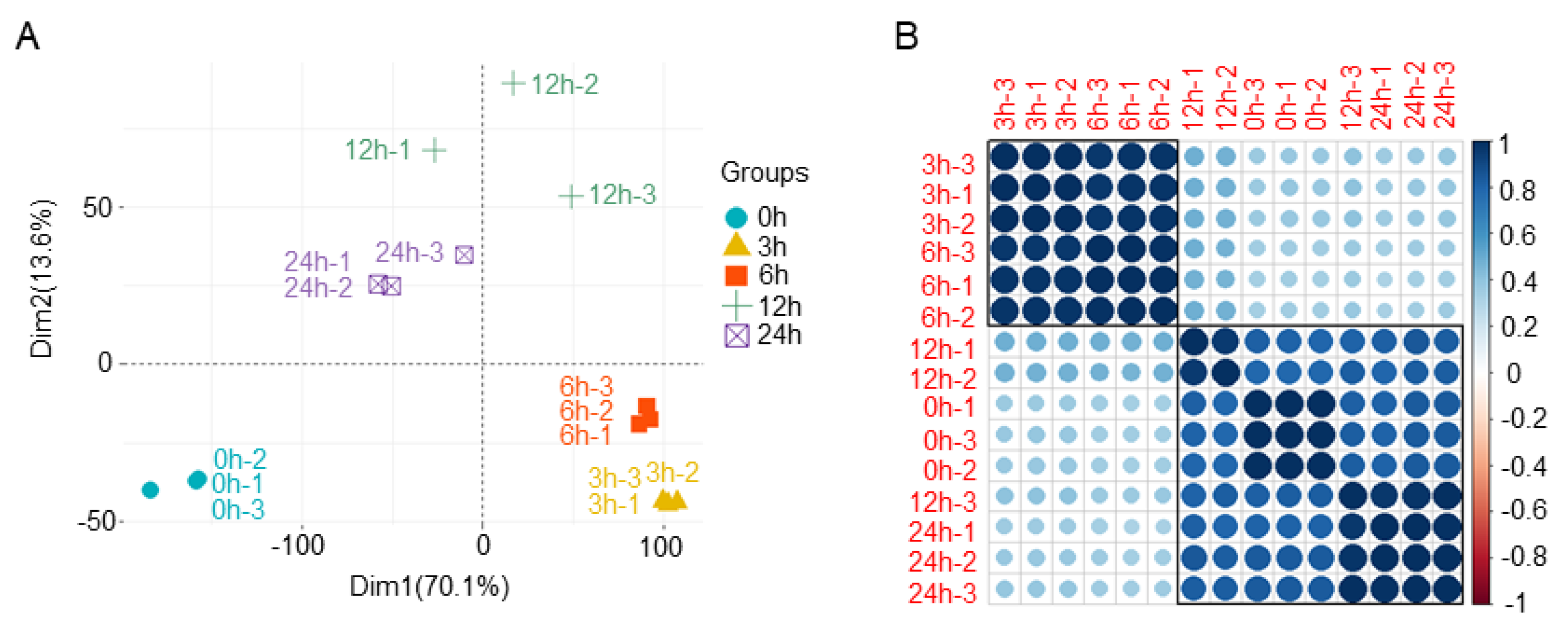
3.1. Evaluation of Transcriptome Sequencing Data
3.2. Differential Gene Expression Analysis
3.3. GO Enrichment Analysis of DEGs
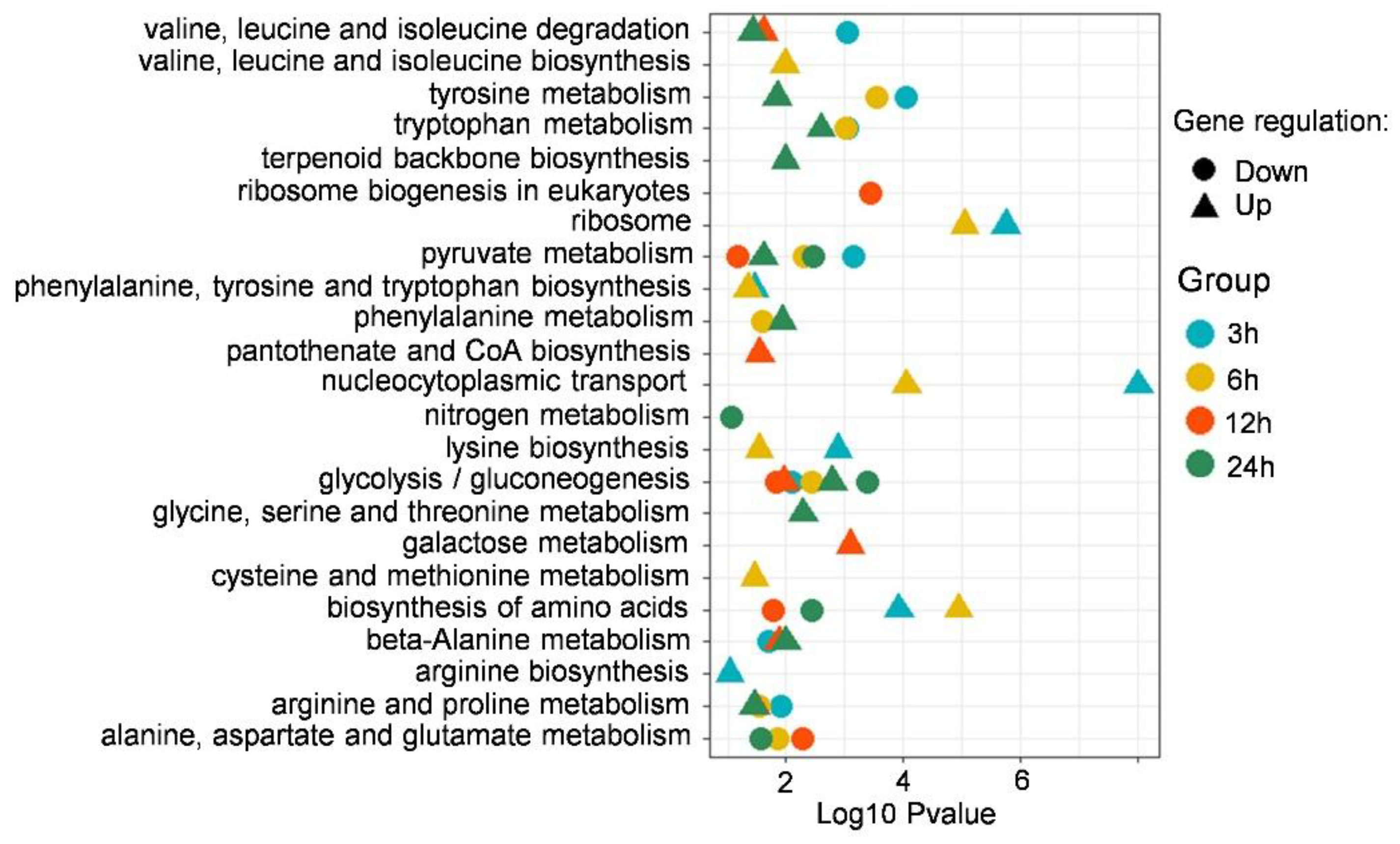
3.4. KEGG Enrichment Analysis of DEGs
3.5. Identification of Differentially Expressed TFs in F. graminearum Treated by Putrescine
3.6. Expression Pattern Analysis of All Genes under Putrescine Treatment
3.7. Construction of Co-Expression Networks
4. Discussion
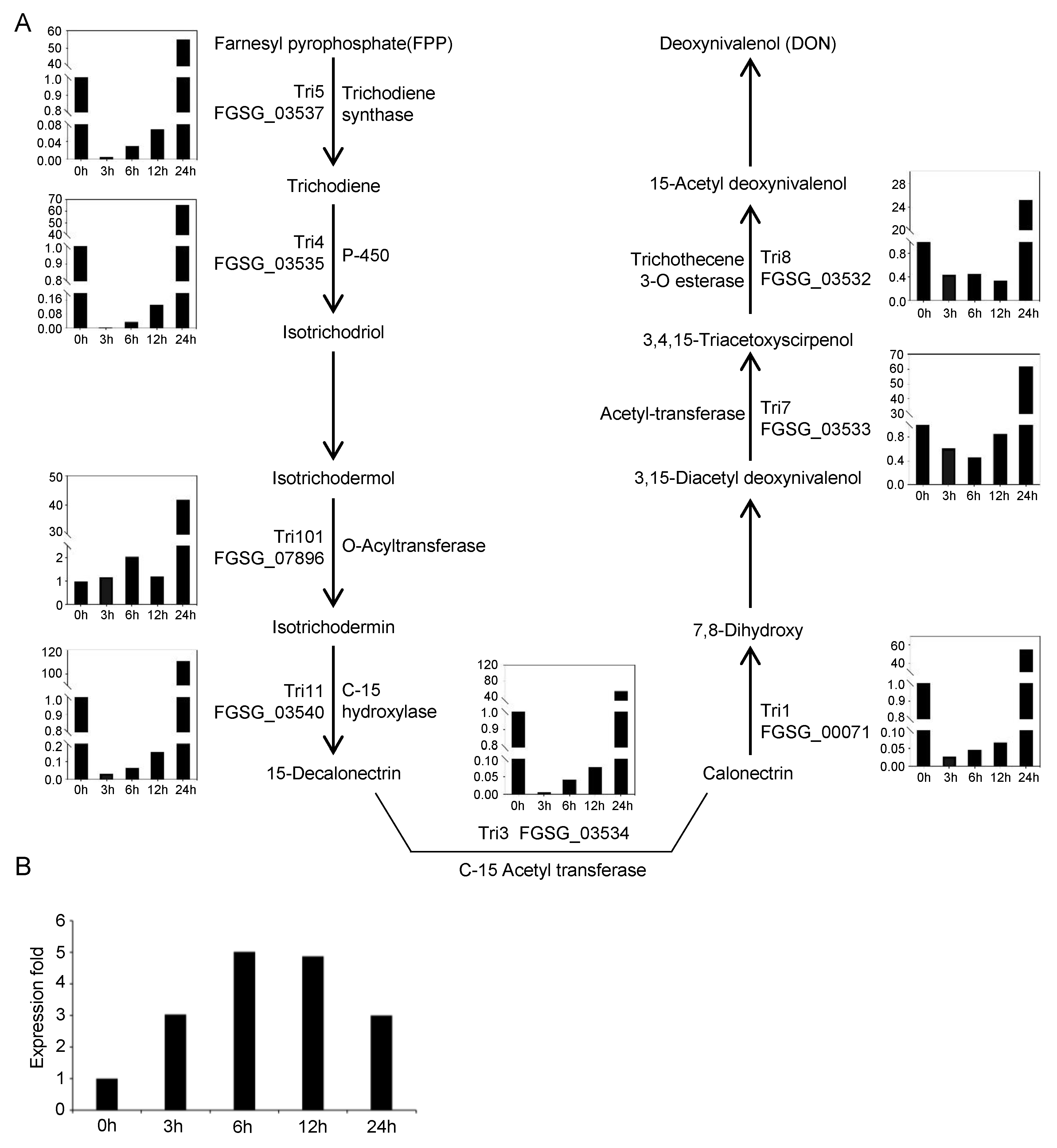
4.1. Expression of FgTRI Genes Began to Increase after 6 h of Putrescine Treatment
4.2. Effect of Amino Acid Biosynthesis and Metabolism on DON Production
4.3. Effect of Glycolysis on DON Production
4.4. FGSG_01915, FGSG_03292, and FGSG_03536 May Be Key TF Genes Involved in DON Synthesis in F. graminearum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, F.; He, D.; Olaniran, A.O.; Mokoena, M.P.; Xu, J.H.; Shi, J.R. Occurrence, toxicity, production and detection of Fusarium mycotoxin: A review. Food Prod. 2019, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.W.; Jenkinson, P.; McLeod, L. Fusarium ear blight (scab) in small grain cereals-a review. Plant Pathol. 1995, 44, 207–238. [Google Scholar] [CrossRef]
- Figueroa, M.; Hammond-Kosack, K.E.; Solomon, P.S. A review of wheat diseases-a field perspective. Mol. Plant Pathol. 2018, 19, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Chakrabortya, S.; Newton, A.C. Climate change, plant diseases and food security: An overview. Plant Pathol. 2011, 60, 2–14. [Google Scholar] [CrossRef]
- West, J.S.; Holdgate, S.; Townsend, J.A.; Edwards, S.G.; Jennings, P.; Fitt, B.D.L. Impacts of changing climate and agronomic factors on fusarium ear blight of wheat in the UK. Fungal Ecol. 2012, 5, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.; Dahl, B.; Nganje, W. Economic costs of Fusarium Head Blight, scab and deoxynivalenol. World Mycotoxin J. 2018, 11, 291–302. [Google Scholar] [CrossRef]
- Khan, M.K.; Pandey, A.; Athar, T.; Choudhary, S.; Deval, R.; Gezgin, S.; Hamurcu, M.; Topal, A.; Atmaca, E.; Santos, P.A. Fusarium head blight in wheat: Contemporary status and molecular approaches. 3 Biotech 2020, 10, 172. [Google Scholar] [CrossRef]
- Xu, F.; Liu, W.; Song, Y.L.; Zhou, Y.L.; Xu, X.M.; Yang, G.Q.; Wang, J.M.; Zhang, J.J.; Liu, L.L. The Distribution of Fusarium graminearum and Fusarium asiaticum Causing Fusarium Head Blight of Wheat in Relation to Climate and Cropping System. Plant Dis. 2021, 105, 2830–2835. [Google Scholar] [CrossRef]
- Kazan, K.; Gardiner, D.M. Transcriptomics of cereal-Fusarium graminearum interactions: What we have learned so far. Mol. Plant Pathol. 2018, 19, 764–778. [Google Scholar] [CrossRef] [Green Version]
- Chilaka, C.A.; Boevre, M.D.; Atanda, O.O.; Saeger, S.D. The Status of Fusarium Mycotoxins in Sub-Saharan Africa: A Review of Emerg ing Trends and Post-Harvest Mitigation Strategies towards Food Control. Toxins 2017, 9, 19. [Google Scholar] [CrossRef]
- Häggblom, P.; Nordkvist, E. Deoxynivalenol, zearalenone, and Fusarium graminearum contamination of cereal straw; field distribution; and sampling of big bales. Mycotoxin Res. 2015, 31, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Kryszczuk, I.P.; Solarska, E.; Wiater, M.K. Communication Reduction of the Fusarium Mycotoxins: Deoxynivalenol, Nivalenol and Zearalenone by Selected Non-Conventional Yeast Strains in Wheat Grains and Bread. Molecules 2022, 27, 1578. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef]
- Liao, Y.X.; Peng, Z.; Chen, L.K.; Nüsslerc, A.K.; Liu, L.G.; Yang, W. Deoxynivalenol, gut microbiota and immunotoxicity: A potential approach? Food Chem. Toxicol. 2018, 112, 342–354. [Google Scholar] [CrossRef]
- Maresca, M. From the gut to the brain: Journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins 2013, 5, 784–820. [Google Scholar] [CrossRef]
- Shifrin, V.I.; Anderson, P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999, 274, 13985–13992. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Ryu, D. Worldwide Occurrence of Mycotoxins in Cereals and Cereal-Derived Food Products: Public Health Perspectives of Their Co-occurrence. J. Agric. Food Chem. 2017, 65, 7034–7051. [Google Scholar] [CrossRef]
- Bian, C.H.; Duan, Y.B.; Xiu, Q.; Wang, J.Y.; Tao, X.; Zhou, M.G. Mechanism of validamycin A inhibiting DON biosynthesis and synergizing with DMI fungicides against Fusarium graminearum. Mol. Plant Pathol. 2021, 22, 769–785. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Kazan, K.; Manners, J.M. Nutrient profiling reveals potent inducers of trichothecene biosynthesis in Fusarium graminearum. Fungal Genet. Biol. 2009, 46, 604–613. [Google Scholar] [CrossRef]
- Gardiner, D.M.; Kazan, K.; Praud, S.; Torney, F.J.; Rusu, A.; Manners, J.M. Early activation of wheat polyamine biosynthesis during Fusarium head blight implicates putrescine as an inducer of trichothecene mycotoxin production. BMC Plant Biol. 2010, 10, 289. [Google Scholar] [CrossRef]
- Crespo-Sempere, A.; Estiarte, N.; Marín, S.; Sanchis, V.; Ramos, A.J. Targeting Fusarium graminearum control via polyamine enzyme inhibitors and polyamine analogs. Food Microbiol. 2015, 49, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.X.; Chen, Y.S.; Shi, C.M.; Huang, Z.B.; Zhang, Y.; Li, S.K.; Li, Y.; Ye, J.; Yu, C.; Li, Z. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among ene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.F.; Zhang, X.L.; Zhao, L.N.; Apaliya, M.T.; Yang, Q.Y.; Sun, W.; Zhang, X.Y.; Zhang, H.Y. Screening of Deoxynivalenol Producing Strains and Elucidation of Possible Toxigenic Molecular Mechanism. Toxins 2017, 9, 184. [Google Scholar] [CrossRef] [Green Version]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Ma, T.L.; Zhang, L.X.; Wang, M.H.; Li, Y.Q.; Jian, Y.Q.; Wu, L.; Kistler, H.C.; Ma, Z.H.; Yin, Y.N. Plant defense compound triggers mycotoxin synthesis by regulating H2B ub1 and H3K4 me2/3 deposition. New Phytol. 2021, 232, 2106–2123. [Google Scholar] [CrossRef]
- McCormick, S.P.; Stanley, A.M.; Stover, N.A.; Alexander, N.J. Trichothecenes: From simple to complex mycotoxins. Toxins 2011, 3, 802–814. [Google Scholar] [CrossRef]
- Chen, Y.; Kistler, H.C.; Ma, Z.H. Fusarium graminearum Trichothecene Mycotoxins: Biosynthesis, Regulation, and Management. Annu. Rev. Phytopathol. 2019, 57, 15–39. [Google Scholar] [CrossRef]
- Alexander, N.J.; Proctor, R.H.; McCormick, S.P. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev. 2009, 28, 198–215. [Google Scholar] [CrossRef]
- Shiobara, T.; Nakajima, Y.; Maeda, K.; Akasaka, M.; Kitou, Y.; Kanamaru, K.; Ohsato, S.; Kobayashi, T.; Nishiuchi, T.; Kimura, M. Identification of amino acids negatively affecting Fusarium trichothecene biosynthesis. Antonie Van Leeuwenhoek 2019, 112, 471–478. [Google Scholar] [CrossRef]
- Giese, H.; Sondergaard, T.E.; Sørensen, J.L. The AreA transcription factor in Fusarium graminearum regulates the use of some nonpreferred nitrogen sources and secondary metabolite production. Fungal Biol. 2013, 117, 814–821. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Li, J.; Duan, Y.B.; Bian, C.H.; Pan, X.Y.; Yao, C.J.; Wang, J.X.; Zhou, M.G. Effects of validamycin in controlling Fusarium head blight caused by Fusarium graminearum: Inhibition of DON biosynthesis and induction of host resistance. Pestic. Biochem. Physiol. 2019, 153, 152–160. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Son, H.; Choi, G.J.; Kim, J.C.; Lee, Y.W. MYT3, a Myb-like transcription factor, affects fungal development and pathogenicity of Fusarium graminearum. PLoS ONE 2014, 9, e94359. [Google Scholar] [CrossRef]
- Shostak, K.; Bonner, C.; Sproule, A.; Thapa, I.; Shields, S.W.J.; Blackwell, B.; Vierula1, J.; Overy, D.; Subramaniam, R. Activation of biosynthetic gene clusters by the global transcriptional regulator TRI6 in Fusarium graminearum. Mol. Microbiol. 2020, 114, 664–680. [Google Scholar] [CrossRef]
- Puri, K.D.; Yan, C.H.; Leng, Y.Q.; Zhong, S.B. RNA-Seq Revealed Differences in Transcriptomes between 3ADON and 15ADON Populations of Fusarium graminearum In Vitro and In Planta. PLoS ONE 2016, 11, e0163803. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clean Data | Mapped Reads | ||||
|---|---|---|---|---|---|---|
| Clean Reads | Q20 (%) | GC (%) | Total Reads | Mapped Reads (%) | Unique Mapped Reads (%) | |
| 0 h | 22,479,084 | 96.12 | 52.54 | 21,130,739 | 20,424,795 (96.66%) | 20,301,372 (96.08%) |
| 0 h | 24,042,002 | 96.45 | 52.50 | 48,283,839 | 46,810,669 (96.95%) | 46,481,429 (96.27%) |
| 0 h | 24,924,258 | 97.07 | 52.43 | 50,074,181 | 48,618,073 (97.09%) | 48,234,367 (96.33%) |
| 3 h | 24,404,822 | 96.98 | 53.83 | 49,178,115 | 47,662,961 (96.92%) | 47,058,097 (95.69%) |
| 3 h | 24,083,702 | 96.12 | 53.82 | 48,540,560 | 46,935,242 (96.69%) | 46,326,950 (95.44%) |
| 3 h | 22,092,868 | 96.14 | 53.73 | 44,533,167 | 43,027,195 (96.62%) | 42,459,736 (95.34%) |
| 6 h | 23,796,099 | 96.94 | 53.88 | 47,915,912 | 46,596,506 (97.25%) | 46,045,762 (96.10%) |
| 6 h | 23,784,079 | 95.86 | 53.75 | 47,893,717 | 46,268,262 (96.61%) | 45,718,258 (95.46%) |
| 6 h | 24,207,815 | 97.06 | 53.12 | 48,760,129 | 47,454,344 (97.32%) | 46,919,058 (96.22%) |
| 12 h | 22,677,537 | 96.69 | 51.18 | 45,602,204 | 44,365,857 (97.29%) | 43,988,722 (96.46%) |
| 12 h | 24,108,403 | 96.28 | 52.57 | 48,460,537 | 46,979,214 (96.94%) | 46,566,522 (96.10%) |
| 12 h | 22,341,753 | 96.19 | 51.91 | 44,895,324 | 43,460,348 (96.80%) | 43,102,193 (96.01%) |
| 24 h | 22,892,156 | 96.48 | 51.26 | 45,998,189 | 44,688,697 (97.15%) | 44,351,292 (96.42%) |
| 24 h | 22,775,413 | 96.33 | 51.52 | 45,736,758 | 44,387,923 (97.05%) | 44,078,173 (96.37%) |
| 24 h | 22,113,432 | 96.09 | 51.70 | 44,418,523 | 43,042,789 (96.90%) | 42,712,534 (96.16%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Zhou, X.; Li, P.; Wang, Y.; Hu, Q.; Shang, Y.; Chen, Y.; Zhu, X.; Feng, H.; Zhang, C. Transcriptome Profile of Fusarium graminearum Treated by Putrescine. J. Fungi 2023, 9, 60. https://doi.org/10.3390/jof9010060
Zhang L, Zhou X, Li P, Wang Y, Hu Q, Shang Y, Chen Y, Zhu X, Feng H, Zhang C. Transcriptome Profile of Fusarium graminearum Treated by Putrescine. Journal of Fungi. 2023; 9(1):60. https://doi.org/10.3390/jof9010060
Chicago/Turabian StyleZhang, Lina, Xishi Zhou, Pengfeng Li, Yiwei Wang, Qianyong Hu, Yuping Shang, Yunshen Chen, Xiying Zhu, Hongjie Feng, and Cuijun Zhang. 2023. "Transcriptome Profile of Fusarium graminearum Treated by Putrescine" Journal of Fungi 9, no. 1: 60. https://doi.org/10.3390/jof9010060