
Transcriptome Profiling Reveals Candidate Genes Related to Stipe Gradient Elongation of Flammulina filiformis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microorganism and Cultivation Conditions
2.2. Stipe Elongation Rate Measurement and Cell Length Detection
2.3. Sample Collection, RNA Extraction, Library Construction, and Sequencing
2.4. RNA-Sequencing Data Analysis
2.5. Gene Identification and Sequence Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Fatty Acid Profile Analysis
2.8. Statistical Analysis
3. Results
3.1. Stipe Gradient Elongation Features of F. filiformis
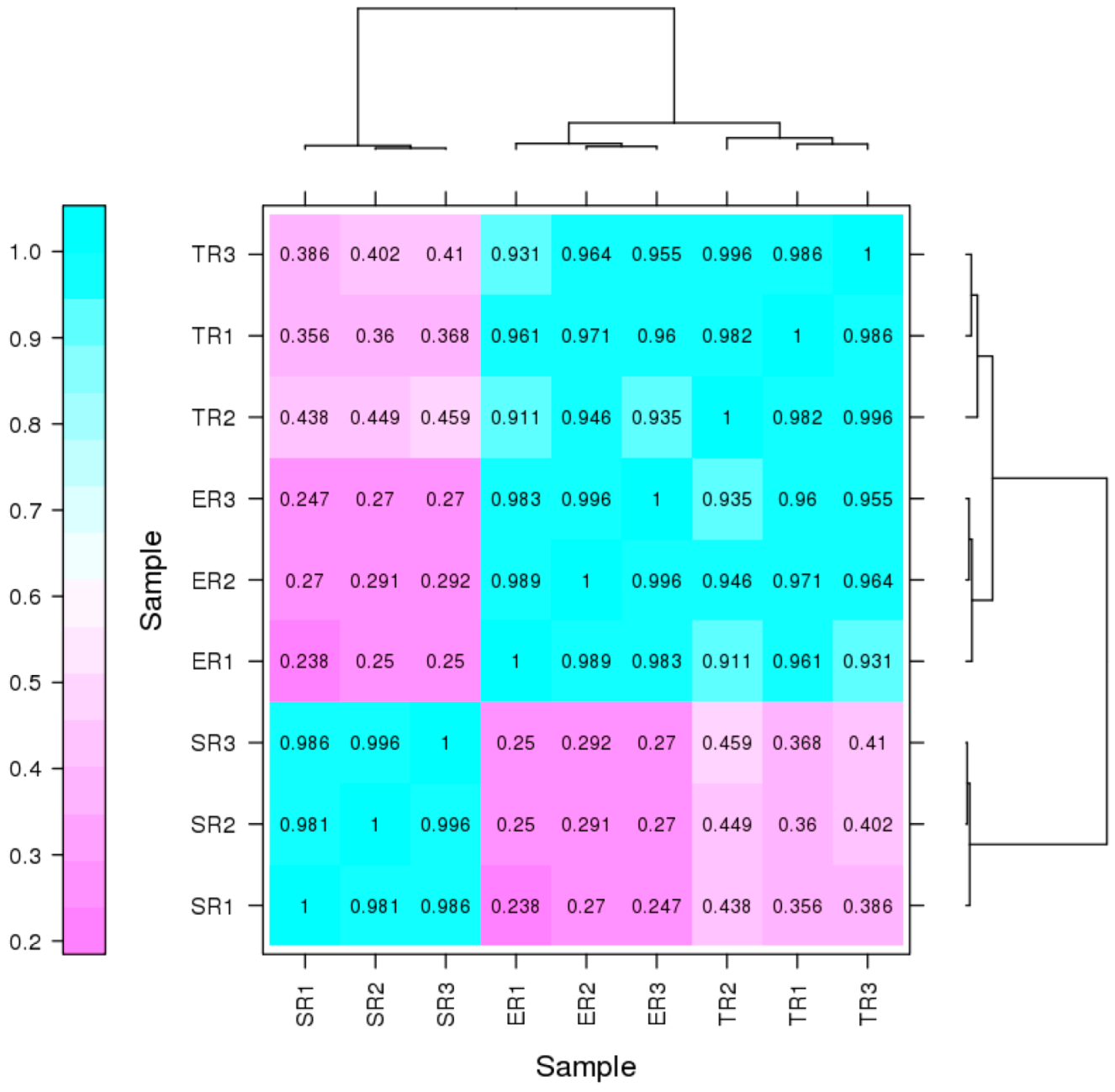
3.2. Transcriptional Changes in Different Stipe Regions of F. filiformis
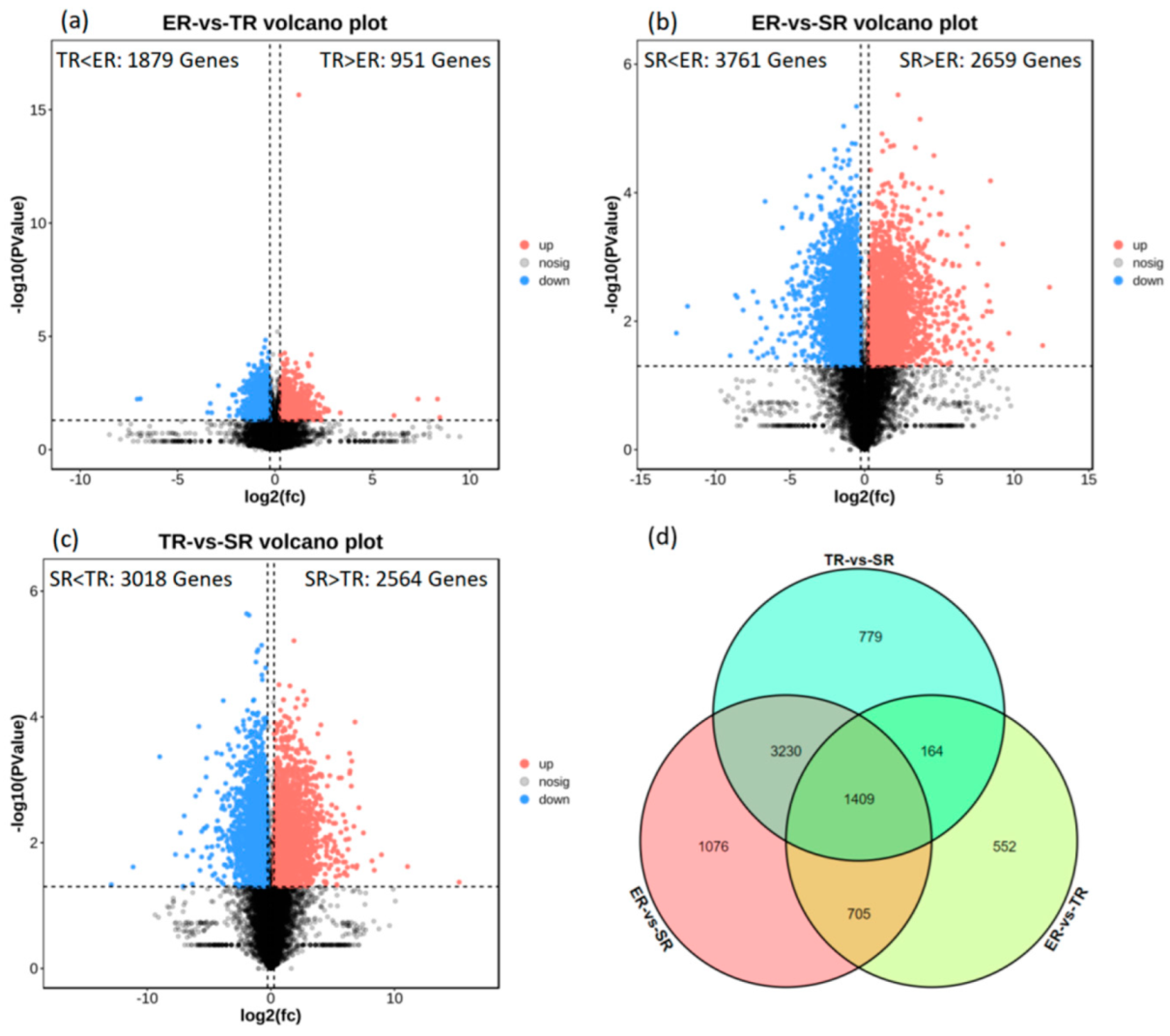
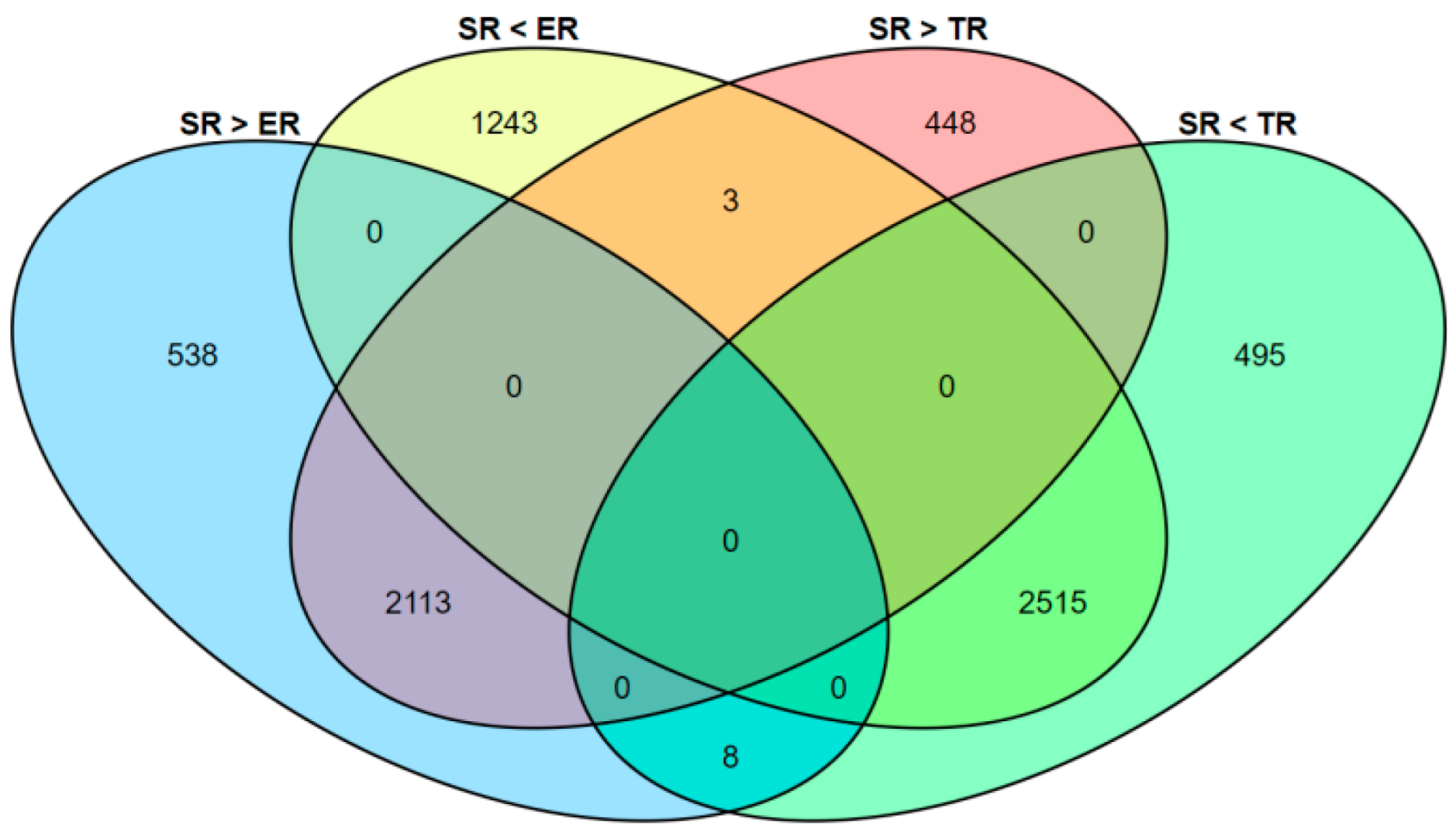
3.3. Differentially Expressed Gene (DEG) Identification
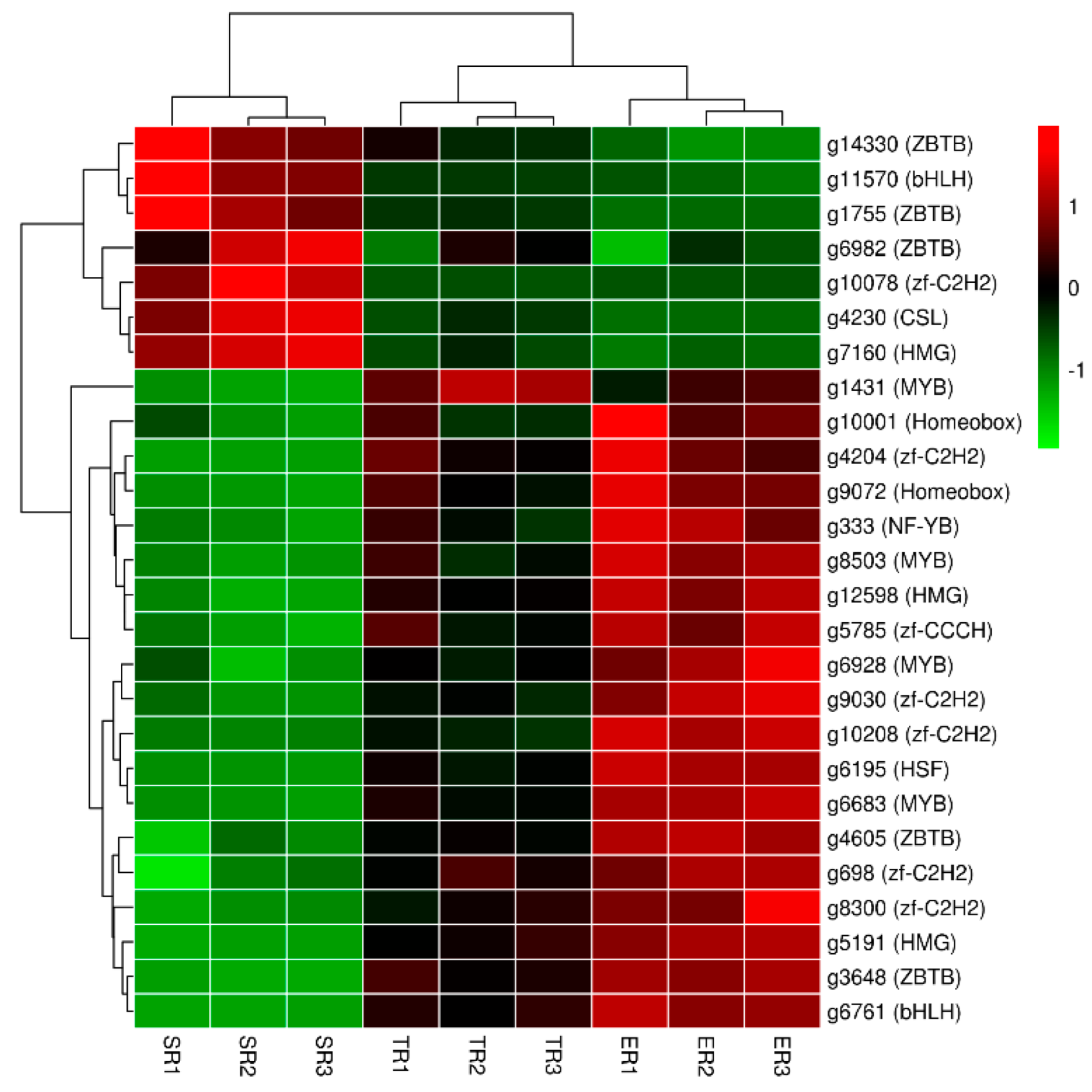
3.4. Transcription Factors (TFs) Annotation of DEGs
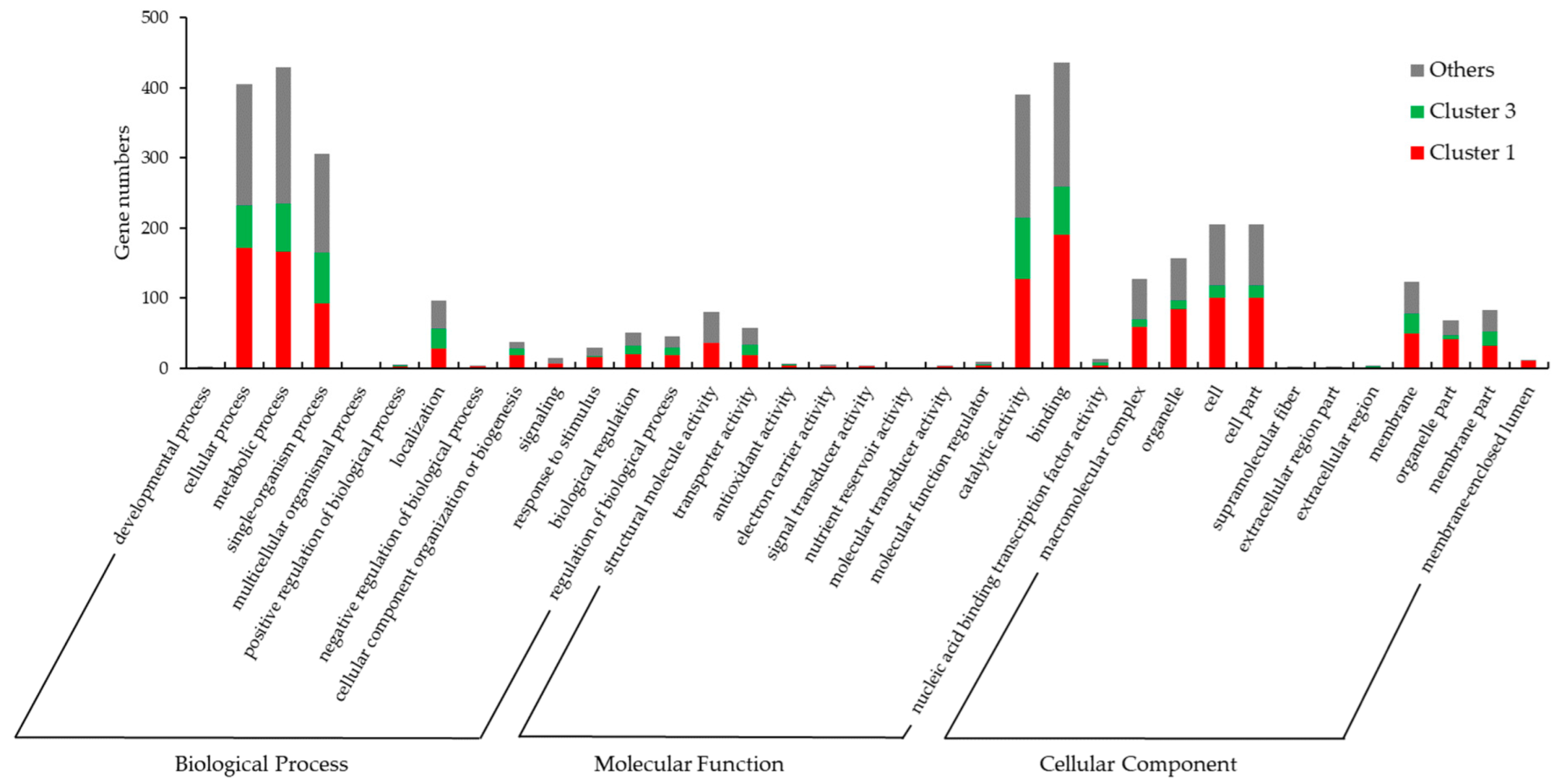
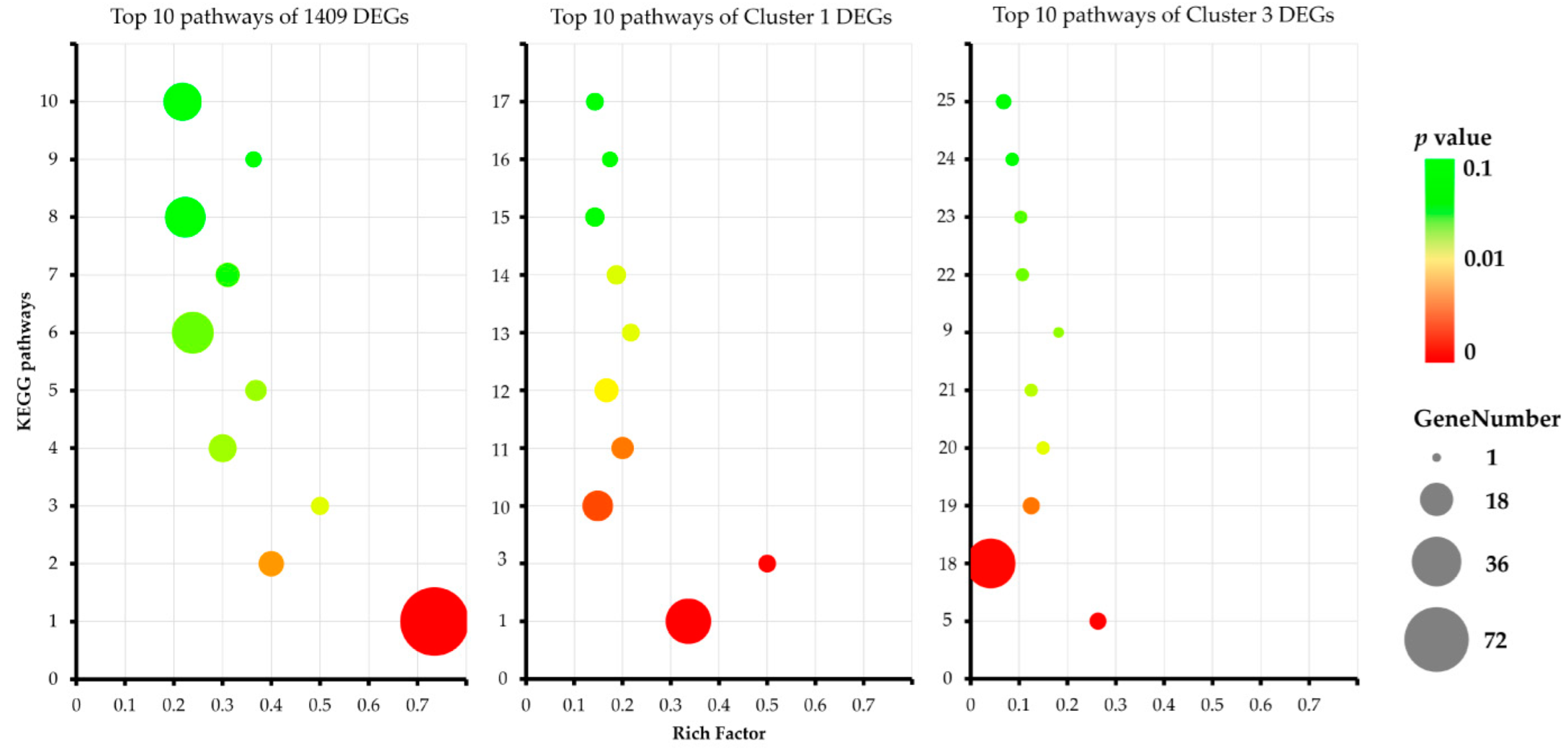
3.5. Gene Ontology (GO) and KEGG Pathway Enrichment Analysis of DEGs
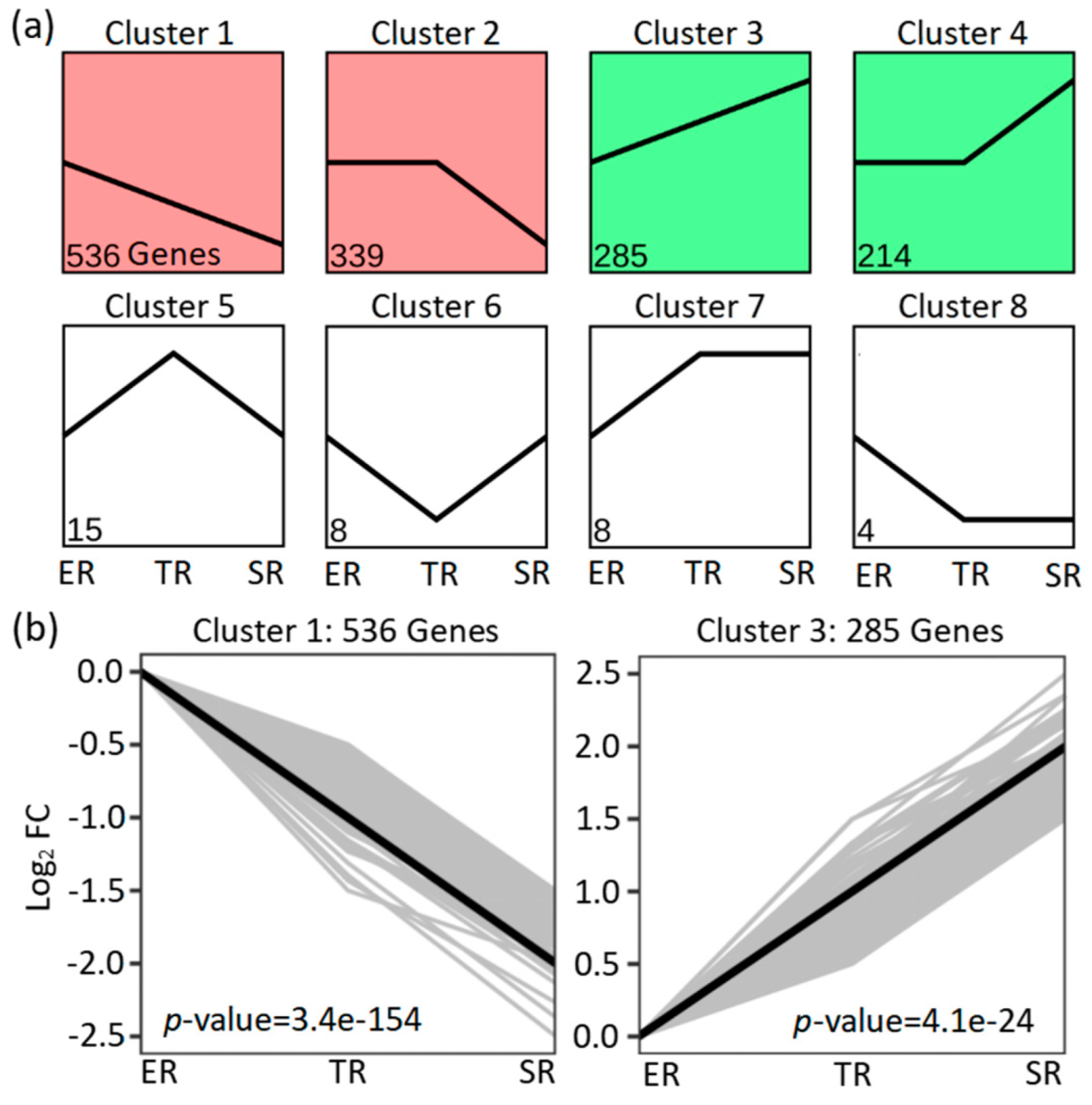
3.6. Ribosome Pathway Gene Expression Patterns Are Consistent with Stipe Elongation Rate
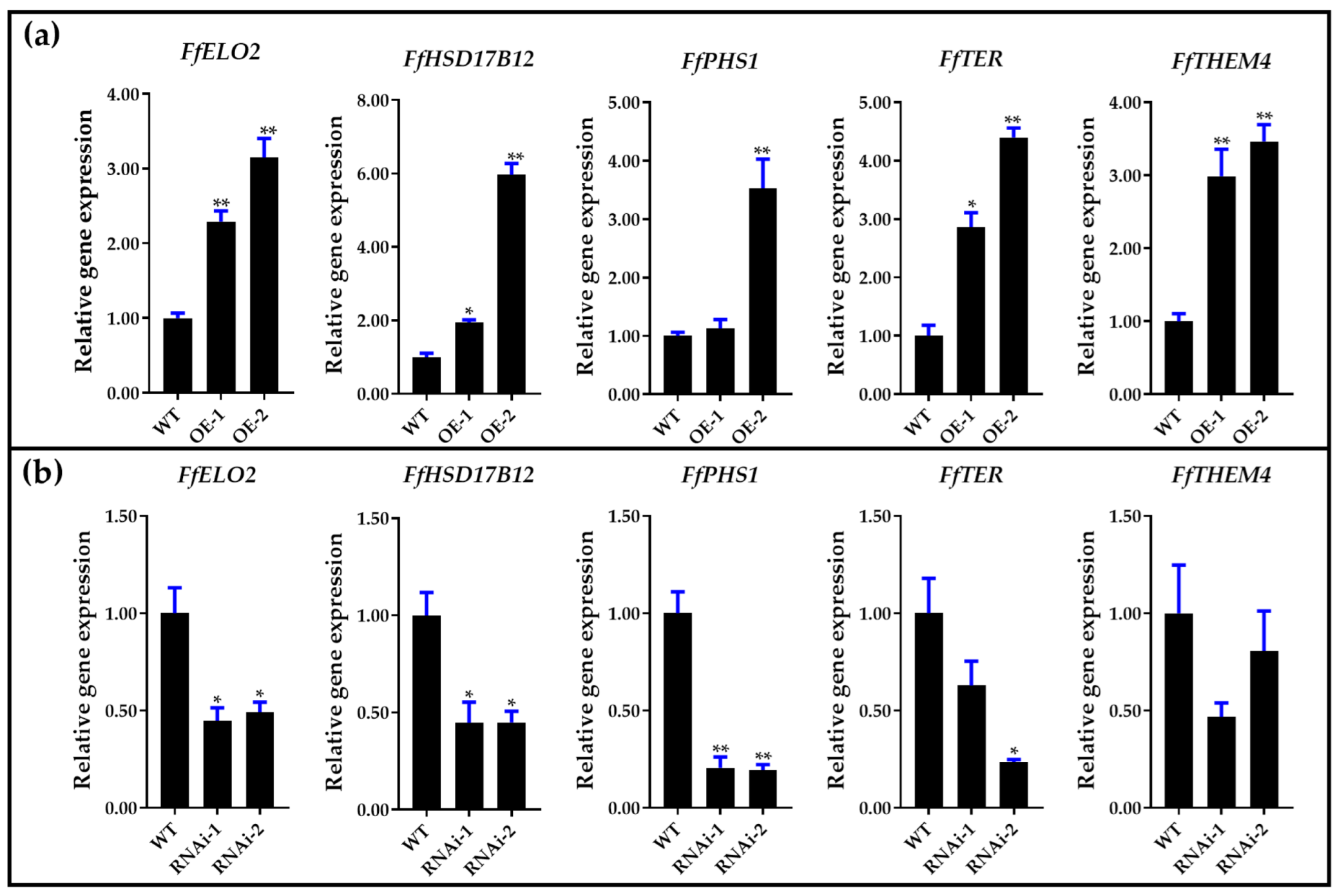
3.7. Long-Chain Fatty Acid Synthesis Pathway Involved in Stipe Gradient Elongation and Regulated by NADPH Oxidase-Derived ROS Signaling Molecules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuees, U.; Navarro-Gonzalez, M. How do Agaricomycetes shape their fruiting bodies? 1. Morphological aspects of development. Fungal Biol. Rev. 2015, 29, 63–97. [Google Scholar] [CrossRef]
- Liu, C.; Bi, J.; Kang, L.; Zhou, J.; Liu, X.; Liu, Z.; Yuan, S. The molecular mechanism of stipe cell wall extension for mushroom stipe elongation growth. Fungal Biol. Rev. 2021, 35, 14–26. [Google Scholar] [CrossRef]
- Craig, G.D.; Gull, K.; Wood, D.A. Stipe elongation in Agaricus bisporus. Microbiology 1977, 102, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wu, X.; Zhou, Y.; Liu, Z.; Zhang, W.; Niu, X.; Zhao, Y.; Pei, S.; Zhao, Y.; Yuan, S. Characterization of stipe elongation of the mushroom Coprinopsis cinerea. Microbiology 2014, 160, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhang, W.; Niu, X.; Liu, Z.; Lu, C.; Wei, H.; Yuan, S. Stipe wall extension of Flammulina velutipes could be induced by an expansin-like protein from Helix aspersa. Fungal Biol. 2014, 118, 1–11. [Google Scholar] [CrossRef]
- Yan, J.; Chekanova, J.; Liu, Y.; Gan, B.; Long, Y.; Han, X.; Tong, Z.; Miao, J.; Lian, L.; Xie, B.; et al. Reactive Oxygen Species Distribution Involved in Stipe Gradient Elongation in the Mushroom Flammulina filiformis. Cells 2022, 11, 1896. [Google Scholar] [CrossRef]
- Kües, U. Life history and developmental processes in the basidiomycete Coprinus cinereus. Microbiol. Mol. Biol. Rev. 2000, 64, 316–353. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Liu, Z.; Zhou, Y.; Wang, J.; Zhang, W.; Yuan, S. Stipe cell wall architecture varies with the stipe elongation of the mushroom Coprinopsis cinerea. Fungal Biol. 2015, 119, 946–956. [Google Scholar] [CrossRef]
- Kang, L.; Zhou, J.; Wang, R.; Zhang, X.; Liu, C.; Liu, Z.; Yuan, S. Glucanase-Induced Stipe Wall Extension Shows Distinct Differences from Chitinase-Induced Stipe Wall Extension of Coprinopsis cinerea. Appl. Env. Microbiol. 2019, 85, e01345-19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kang, L.; Liu, C.; Niu, X.; Wang, X.; Liu, H.; Zhang, W.; Liu, Z.; Latgé, J.P.; Yuan, S. Chitinases play a key role in stipe cell wall extension in the mushroom Coprinopsis cinerea. Appl. Environ. Microbiol. 2019, 85, e00532-19. [Google Scholar] [CrossRef]
- Kang, L.; Zhang, X.; Liu, X.; Wang, R.; Liu, C.; Zhou, J.; Liu, Z.; Yuan, S. Comparative study of β-glucan-degrading enzymes from Coprinopsis cinerea for their capacities to induce stipe cell wall extension. Int. J. Biol. Macromol. 2020, 152, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Bi, J.; Bai, Y.; Duan, B.; Liu, Z.; Yuan, S. Accumulation and cross-linkage of β-1, 3/1, 6-glucan lead to loss of basal stipe cell wall extensibility in mushroom Coprinopsis cinerea. Carbohydr. Polym. 2021, 259, 117743. [Google Scholar] [CrossRef]
- Shioya, T.; Nakamura, H.; Ishii, N.; Takahashi, N.; Sakamoto, Y.; Ozaki, N.; Kobayashi, M.; Okano, K.; Kamada, T.; Muraguchi, H. The Coprinopsis cinerea septin Cc.Cdc3 is involved in stipe cell elongation. Fungal Genet. Biol. 2013, 58, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yuan, J.; Chen, Y.; Wang, Y.; Chen, J.; Bi, J.; Lyu, L.; Yu, C.; Yuan, S.; Liu, Z. MAPK CcSakA of the HOG Pathway Is Involved in Stipe Elongation during Fruiting Body Development in Coprinopsis cinerea. J. Fungi 2022, 8, 534. [Google Scholar] [CrossRef]
- Huang, Q.; Han, X.; Mukhtar, I.; Gao, L.; Huang, R.; Fu, L.; Yan, J.; Tao, Y.; Chen, B.; Xie, B. Identification and expression patterns of fvexpl1, an expansin-like protein-encoding gene, suggest an auxiliary role in the stipe morphogenesis of Flammulina velutipes. J. Microbiol. Biotechnol. 2018, 28, 622–629. [Google Scholar] [CrossRef]
- Huang, Q.; Mukhtar, I.; Zhang, Y.; Wei, Z.; Han, X.; Huang, R.; Yan, J.; Xie, B. Identification and characterization of two new s-adenosylmethionine-dependent methyltransferase encoding genes suggested their involvement in stipe elongation of Flammulina velutipes. Mycobiology 2019, 47, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.Q.; Yan, J.J.; Li, Y.N.; Yang, H.; Ma, X.B.; Wang, M.; Tao, Y.X.; Xie, B.G. Cytochrome c peroxidase gene (ffccp) and its differential expression during stipe elongation in Flammulina filiformis. Mycosystema 2020, 39, 993–1005. [Google Scholar]
- Li, J.; Shao, Y.; Yang, Y.; Xu, C.; Jing, Z.; Li, H.; Xie, B.; Tao, Y. The chromatin modifier protein FfJMHY plays an important role in regulating the rate of mycelial growth and stipe elongation in Flammulina filiformis. J. Fungi 2022, 8, 477. [Google Scholar] [CrossRef]
- Nowrousian, M. Genomics and transcriptomics to study fruiting body development: An update. Fungal Biol. Rev. 2018, 32, 231–235. [Google Scholar] [CrossRef]
- Tao, Y.; Van Peer, A.F.; Chen, B.; Chen, Z.; Zhu, J.; Deng, Y.; Jiang, Y.; Li, S.; Wu, T.; Xie, B. Gene expression profiling reveals large regulatory switches between succeeding stipe stages in Volvariella volvacea. PLoS ONE 2014, 9, e97789. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.B.; Xia, E.H.; Li, M.; Cui, Y.Y.; Wang, P.M.; Zhang, J.X.; Xie, B.G.; Xu, J.P.; Yan, J.J.; Li, J.; et al. Transcriptome data reveal conserved patterns of fruiting body development and response to heat stress in the mushroom-forming fungus Flammulina filiformis. PLoS ONE 2020, 15, e0239890. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.J.; Tong, Z.J.; Liu, Y.Y.; Li, Y.N.; Zhao, C.; Mukhtar, I.; Tao, Y.X.; Chen, B.Z.; Deng, Y.J.; Xie, B.G. Comparative transcriptomics of Flammulina filiformis suggests a high CO2 concentration inhibits early pileus expansion by decreasing cell division control pathways. Int. J. Mol. Sci. 2019, 20, 5923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Ding, X.; Hou, Y. Comparative analysis of transcriptomes revealed the molecular mechanism of development of Tricholoma matsutake at different stages of fruiting bodies. Food Sci. Biotechnol. 2020, 29, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gong, W.; Li, C.; Shen, N.; Gui, Y.; Bian, Y.; Kwan, H.S.; Cheung, M.K.; Xiao, Y. RNA-Seq-based high-resolution linkage map reveals the genetic architecture of fruiting body development in shiitake mushroom, Lentinula edodes. Comput. Struct. Biotechnol. J. 2021, 19, 1641–1653. [Google Scholar] [CrossRef]
- Orban, A.; Weber, A.; Herzog, R.; Hennicke, F.; Rühl, M. Transcriptome of different fruiting stages in the cultivated mushroom Cyclocybe aegerita suggests a complex regulation of fruiting and reveals enzymes putatively involved in fungal oxylipin biosynthesis. BMC Genom. 2021, 22, 324. [Google Scholar] [CrossRef]
- Liu, D.; Sun, X.; Diao, W.; Qi, X.; Bai, Y.; Yu, X.; Li, L.; Fang, H.; Chen, Z.; Liu, Q.; et al. Comparative transcriptome analysis revealed candidate genes involved in fruiting body development and sporulation in Ganoderma lucidum. Arch. Microbiol. 2022, 204, 514. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.M.; Tang, Y.J.; Ma, K.; Li, B.; Zeng, X.; Liu, X.B.; Li, Y.; Yang, Z.L.; Xu, W.N.; et al. Genome-wide analysis and prediction of genes involved in the biosynthesis of polysaccharides and bioactive secondary metabolites in high-temperature-tolerant wild Flammulina filiformis. BMC Genom. 2020, 21, 719. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.-J.; Zhang, L.; Wang, R.-Q.; Xie, B.; Li, X.; Chen, R.-L.; Guo, L.X.; Xie, B.G. The sequence characteristics and expression models reveal superoxide dismutase involved in cold response and fruiting body development in Volvariella volvacea. Int. J. Mol. Sci. 2016, 17, 34. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Van Peer, A.F.; Huang, Q.; Shao, Y.; Zhang, L.; Xie, B.; Jiang, Y.; Zhu, J.; Xie, B. Identification of novel and robust internal control genes from Volvariella volvacea that are suitable for RT-qPCR in filamentous fungi. Sci. Rep. 2016, 6, 29236. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, Y.; Zhuang, Y.; Qin, Y.; Sun, L. Nonvolatile taste components, nutritional values, bioactive compounds and antioxidant activities of three wild Chanterelle mushrooms. Int. J. Food Sci. Technol. 2018, 53, 1855–1864. [Google Scholar] [CrossRef]
- Pelkmans, J.F.; Patil, M.B.; Gehrmann, T.; Reinders, M.J.; Wösten, H.A.; Lugones, L.G. Transcription factors of Schizophyllum commune involved in mushroom formation and modulation of vegetative growth. Sci. Rep. 2017, 7, 310. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.E. A role for the ribosome in development. Trends Plant Sci. 2009, 14, 512–519. [Google Scholar] [CrossRef]
- Norris, K.; Hopes, T.; Aspden, J.L. Ribosome heterogeneity and specialization in development. Wiley Interdiscip. Rev. RNA 2021, 12, e1644. [Google Scholar] [CrossRef]
- Qin, Y.M.; Hu, C.Y.; Pang, Y.; Kastaniotis, A.J.; Hiltunen, J.K.; Zhu, Y.X. Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 2007, 19, 3692–3704. [Google Scholar] [CrossRef] [Green Version]
- Kniazeva, M.; Sieber, M.; McCauley, S.; Zhang, K.; Watts, J.L.; Han, M. Suppression of the ELO-2 FA elongation activity results in alterations of the fatty acid composition and multiple physiological defects, including abnormal ultradian rhythms, in Caenorhabditis elegans. Genetics 2003, 163, 159–169. [Google Scholar] [CrossRef]
- Piszczatowska, K.; Przybylska, D.; Sikora, E.; Mosieniak, G. Inhibition of NADPH oxidases activity by diphenyleneiodonium chloride as a mechanism of senescence induction in human cancer cells. Antioxidants 2020, 9, 1248. [Google Scholar] [CrossRef]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef] [PubMed]
- Muraguchi, H.; Umezawa, K.; Niikura, M.; Yoshida, M.; Kozaki, T.; Ishii, K.; Sakai, K.; Shimizu, M.; Nakahori, K.; Sakamoto, Y.; et al. Strand-specific RNA-seq analyses of fruiting body development in Coprinopsis cinerea. PLoS ONE 2015, 10, e0141586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, T. Stipe elongation in fruit bodies. In Growth, Differentiation and Sexuality; Wessels, J.G., Meinhardt, F., Eds.; Springer: Heidelberg, Germany, 1994; pp. 367–379. [Google Scholar]
- Mau, J.L.; Beelman, R.B.; Ziegler, G.R. Effect of 10-oxo-trans-8-decenoic acid on growth of Agaricus bisporus. Phytochemistry 1992, 31, 4059–4064. [Google Scholar] [CrossRef]
- Champavier, Y.; Pommier, M.T.; Arpin, N.; Voiland, A.; Pellon, G. 10-Oxo-trans-8-decenoic acid (ODA): Production, biological activities, and comparison with other hormone-like substances in Agaricus bisporus. Enzym. Microb. Technol. 2000, 26, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Mau, J.L.; Ma, J.T. Effect of 10-oxo-trans-8-decenoic acid on growth of several mushroom mycelia. Fungi Sci. 2001, 16, 1–12. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Tong, Z.; Han, X.; Gan, Y.; Liu, Y.; Chen, J.; Duan, X.; Lin, J.; Gan, B.; Xie, B. Transcriptome Profiling Reveals Candidate Genes Related to Stipe Gradient Elongation of Flammulina filiformis. J. Fungi 2023, 9, 64. https://doi.org/10.3390/jof9010064
Yan J, Tong Z, Han X, Gan Y, Liu Y, Chen J, Duan X, Lin J, Gan B, Xie B. Transcriptome Profiling Reveals Candidate Genes Related to Stipe Gradient Elongation of Flammulina filiformis. Journal of Fungi. 2023; 9(1):64. https://doi.org/10.3390/jof9010064
Chicago/Turabian StyleYan, Junjie, Zongjun Tong, Xing Han, Ying Gan, Yuanyuan Liu, Jie Chen, Xinlian Duan, Junbin Lin, Bingcheng Gan, and Baogui Xie. 2023. "Transcriptome Profiling Reveals Candidate Genes Related to Stipe Gradient Elongation of Flammulina filiformis" Journal of Fungi 9, no. 1: 64. https://doi.org/10.3390/jof9010064