
Characterization of Complete Mitochondrial Genomes of the Five Peltigera and Comparative Analysis with Relative Species


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequencing, Assembly, and Annotation of Mitochondrial Genomes
2.3. Sequence Analyses of Mitogenomes
2.4. Repetitive Element Analysis
2.5. Phylogenetic Analysis
3. Results
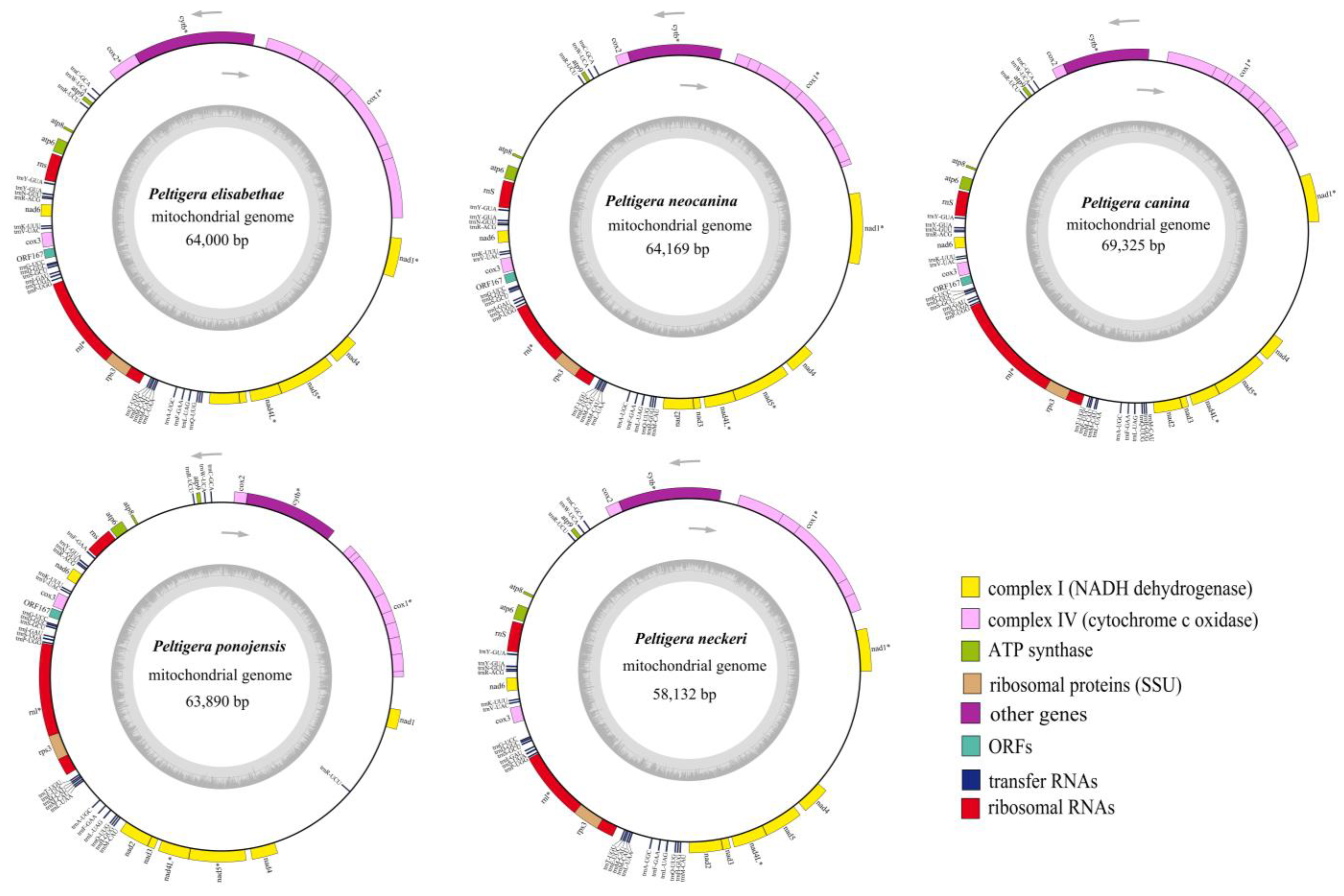
3.1. Mitogenome Features
3.2. RNA Genes
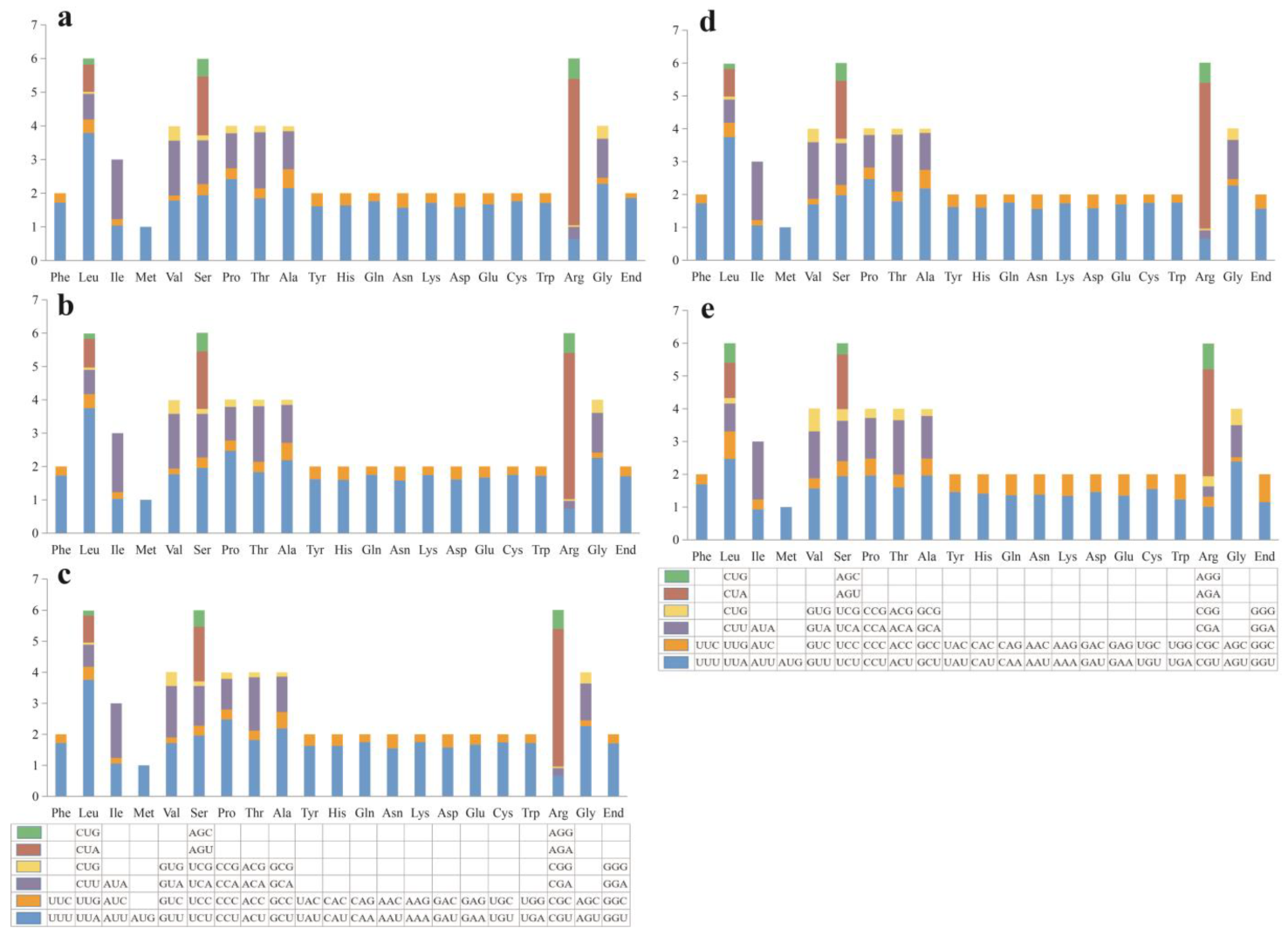
3.3. Codon Usage Analysis
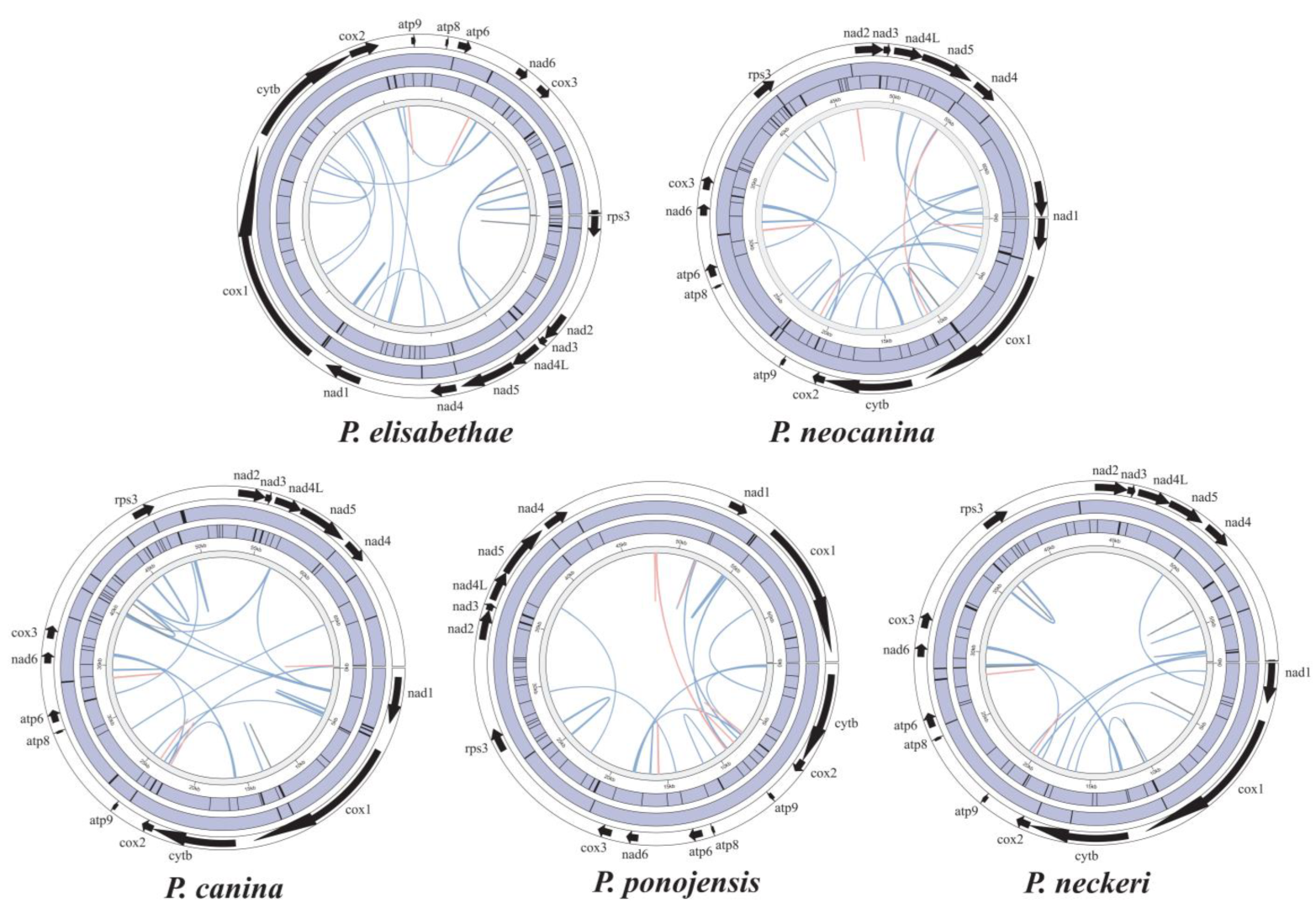
3.4. Repetitive Element Analysis
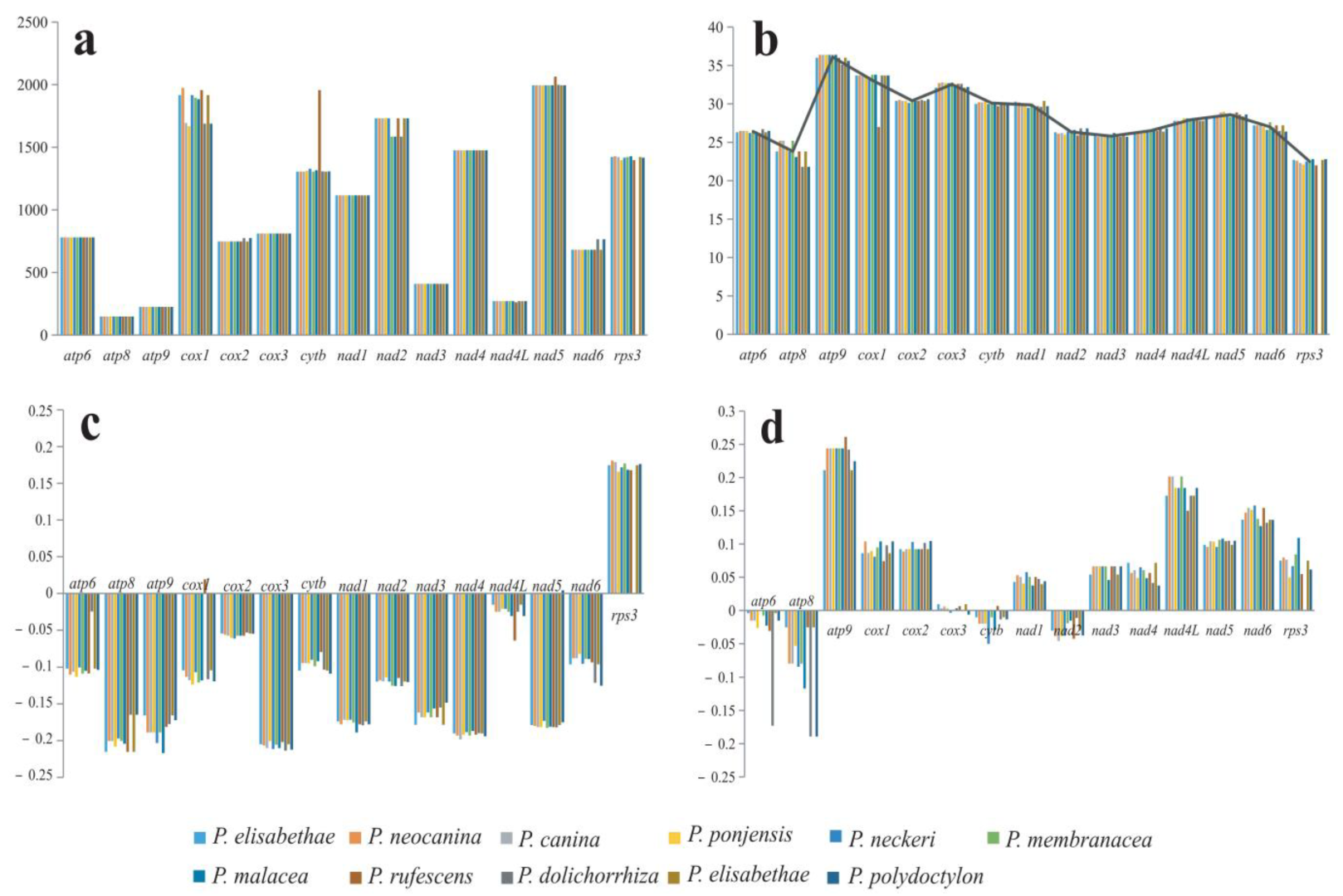
3.5. Variation, Genetic Distance, and Evolutionary Rates of PCGs
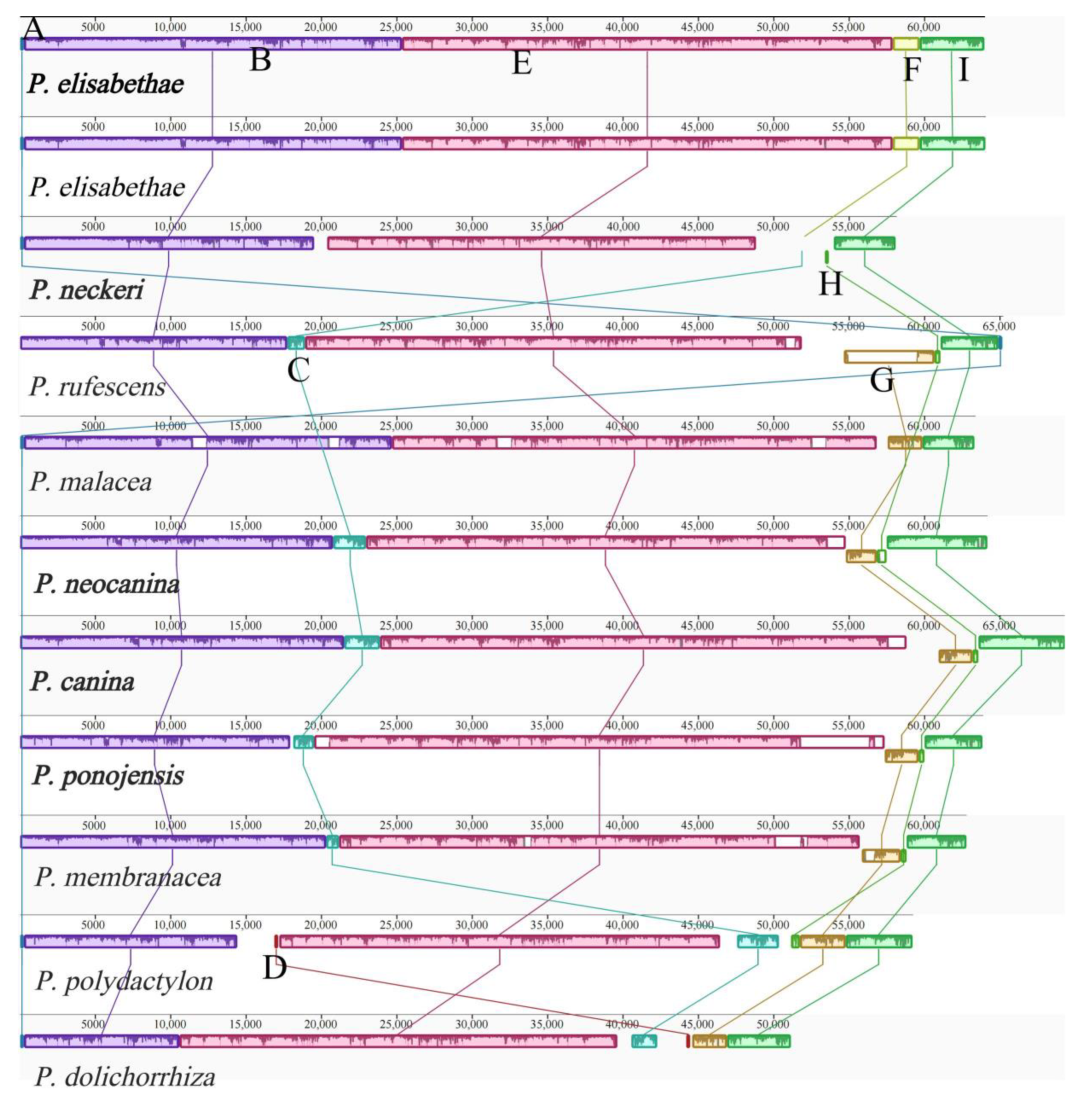
3.6. Synteny Analysis
3.7. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ahmadjian, V. The Lichen Symbiosis; John Wiley and Sons: New York, NY, USA, 1993. [Google Scholar]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; McCutcheon, J.P. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef]
- Sharma, M.; Mohammad, A. Lichens and lichenology: Historical and economic prospects. Lichen-Deriv. Prod. Extr. Appl. 2020, 101–118. [Google Scholar] [CrossRef]
- Watkinson, S.C. Mutualistic symbiosis between fungi and autotrophs. In The Fungi; Academic Press: Cambridge, MA, USA, 2016; pp. 205–243. [Google Scholar]
- Honegger, R. The lichen symbiosis—What is so spectacular about it? Lichenologist 1998, 30, 193–212. [Google Scholar] [CrossRef]
- Kappen, L. Response to extreme environments. In The Lichens; Academic Press: Cambridge, MA, USA, 1973; pp. 311–380. [Google Scholar]
- Shukla, V.; Kumar, S.; Kumar, N. Plant Adaptation Strategies in Changing Environment; Springer: Singapore, 2017. [Google Scholar]
- Kłos, A.; Rajfur, M.; Šrámek, I.; Wacławek, M. Use of lichen and moss in assessment of forest contamination with heavy metals in Praded and Glacensis Euroregions (Poland and Czech Republic). Water Air Soil Pollut. 2011, 222, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, M.; Xu, B. A comprehensive review on secondary metabolites and health-promoting effects of edible lichen. J. Funct. Foods 2021, 80, 104283. [Google Scholar] [CrossRef]
- Deduke, C.; Timsina, B.; Piercey-Normore, M.D. Effect of environmental change on secondary metabolite production in lichen-forming fungi. In International Perspectives on Global Environmental Change; InTech: Houston, TX, USA, 2012; pp. 197–230. [Google Scholar]
- Luo, H.; Wei, X.; Yamamoto, Y.; Liu, Y.; Wang, L.; Jung, J.S.; Hur, J.S. Antioxidant activities of edible lichen Ramalina conduplicans and its free radical-scavenging constituents. Mycoscience 2010, 51, 391–395. [Google Scholar] [CrossRef]
- Honegger, R. Functional aspects of the lichen symbiosis. Annu. Rev. Plant Biol. 1991, 42, 553–578. [Google Scholar] [CrossRef]
- MartÍnez, I.; Burga, A.R.; Vitikainen, O.; Escudero, A. Distribution patterns in the genus Peltigera Willd. Lichenologist 2003, 35, 301–323. [Google Scholar] [CrossRef]
- Miadlikowska, J.; Lutzoni, F. Phylogenetic revision of the genus Peltigera (lichen-forming Ascomycota) based on morphological, chemical, and large subunit nuclear ribosomal DNA data. Int. J. Plant Sci. 2000, 161, 925–958. [Google Scholar] [CrossRef]
- Magain, N.; Tniong, C.; Goward, T.; Niu, D.; Goffinet, B.; Sérusiaux, E.; Miadlikowska, J. Species delimitation at a global scale reveals high species richness with complex biogeography and patterns of symbiont association in Peltigera section Peltigera (lichenized Ascomycota: Lecanoromycetes). Taxon 2018, 67, 836–870. [Google Scholar] [CrossRef]
- Miadlikowska, J.; Richardson, D.; Magain, N. Phylogenetic placement, species delimitation, and cyanobiont identity of endangered aquatic Peltigera species (lichen-forming Ascomycota, Lecanoromycetes). Am. J. Bot. 2014, 101, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Miadlikowska, J.; Magain, N.; Pardo de la Hoz, C.; Niu, D.; Goward, T.; Sérusiaux, E.; Lutzoni, F. Species in section Peltidea (aphthosa group) of the genus Peltigera remain cryptic after molecular phylogenetic revision. Plant Fungal Syst. 2018, 63, 45–64. [Google Scholar] [CrossRef]
- Funk, E.R.; Adams, A.N.; Spotten, S.M.; Van Hove, R.A.; Whittington, K.T.; Keepers, K.G.; Kane, N.C. The complete mitochondrial genomes of five lichenized fungi in the genus Usnea (Ascomycota: Parmeliaceae). Mitochondrial DNA Part B 2018, 3, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.; Liu, Y.; Sérusiaux, E.; Goffinet, B. Complete mitogenome sequence of Ricasolia amplissima (Lobariaceae) reveals extensive mitochondrial DNA rearrangement within the Peltigerales (lichenized ascomycetes). Bryologist 2017, 120, 335–339. [Google Scholar] [CrossRef]
- Lan, Y.; Huang, F. The complete mitochondrial genome of the lichenized fungi Usnea jiangxiensis (Ascomycota: Parmeliaceae). Mitochondrial DNA Part B 2020, 5, 1477–1478. [Google Scholar] [CrossRef]
- Magain, N.; Miadlikowska, J.; Goffinet, B.; Sérusiaux, E.; Lutzoni, F. Macroevolution of specificity in cyanolichens of the genus Peltigera section Polydactylon (Lecanoromycetes, Ascomycota). Syst. Biol. 2017, 66, 74–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mamut, R. Mitochondrial genome from the lichenized fungus Peltigera rufescens (Weiss) Humb, 1793 (Ascomycota: Peltigeraceae). Mitochondrial DNA Part B 2021, 6, 2186–2187. [Google Scholar] [CrossRef]
- Xavier, B.B.; Miao, V.P.; Jonsson, Z.O.; Andresson, O.S. Mitochondrial genomes from the lichenized fungi Peltigera membranacea and Peltigera malacea: Features and phylogeny. Fungal Biol. 2012, 116, 802–814. [Google Scholar] [CrossRef]
- Delsuc, F.; Stanhope, M.J.; Douzery, E.J. Molecular systematics of armadillos (Xenarthra, Dasypodidae): Contribution of maximum likelihood and Bayesian analyses of mitochondrial and nuclear genes. Mol. Phylogenet. Evol. 2003, 28, 261–275. [Google Scholar] [CrossRef]
- Hassanin, A.; An, J.; Ropiquet, A.; Nguyen, T.T.; Couloux, A. Combining multiple autosomal introns for studying shallow phylogeny and taxonomy of Laurasiatherian mammals: Application to the tribe Bovini (Cetartiodactyla, Bovidae). Mol. Phylogenet. Evol. 2013, 66, 766–775. [Google Scholar] [CrossRef]
- O’Brien, H.E.; Miadlikowska, J.; Lutzoni, F. Assessing reproductive isolation in highly diverse communities of the lichen-forming fungal genus Peltigera. Evolution 2009, 63, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. Royal. Soc. B 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Nelsen, M.P.; Lücking, R.; Grube, M.; Mbatchou, J.S.; Muggia, L.U.C.I.A.; Plata, E.R.; Lumbsch, H.T. Unravelling the phylogenetic relationships of lichenised fungi in Dothideomyceta. Stud. Mycol. 2009, 64, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Carpi, G.; Kitchen, A.; Kim, H.L.; Ratan, A.; Drautz-Moses, D.I.; McGraw, J.J.; Schuster, S.C. Mitogenomes reveal diversity of the European Lyme borreliosis vector Ixodes ricinus in Italy. Mol. Biol. Evol. 2016, 101, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.; Sidore, C.; Achilli, A.; Angius, A.; Posth, C.; Furtwängler, A.; Torroni, A. Mitogenome diversity in Sardinians: A genetic window onto an island’s past. Mol. Biol. Evol. 2017, 34, 1230–1239. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed]
- Valach, M.; Burger, G.; Gray, M.; Lang, B.F. Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucleic Acids Res. 2014, 42, 13764–13777. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Andrzej, E.; Jim, O. The Bacterial, Archaeal and Plant Plastid Code. 2013. [Google Scholar]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Q.; Jin, X.; Chen, Z.; Xiong, C.; Li, P.; Huang, W. Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. J. Biol. Macromol. 2019, 121, 249–260. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Xiang, C.Y.; Gao, F.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.L.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Li, Q.; Wu, P.; Li, L.; Feng, H.; Tu, W.; Bao, Z.; Huang, W. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 2021, 172, 560–572. [Google Scholar] [CrossRef]
- Fonseca, P.L.; De-Paula, R.B.; Araújo, D.S.; Tomé, L.M.R.; Mendes-Pereira, T.; Rodrigues, W.F.C.; Góes-Neto, A. Global characterization of fungal mitogenomes: New insights on genomic diversity and dynamism of coding genes and accessory elements. Front. Microbiol. 2021, 12, 787283. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhang, G.; Liu, X.; Wang, C.; Xu, J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 2015, 77, 95–107. [Google Scholar] [CrossRef]
- Blakely, E.L.; Yarham, J.W.; Alston, C.L.; Craig, K.; Poulton, J.; Brierley, C.; Taylor, R.W. Pathogenic Mitochondrial t RNA Point Mutations: Nine Novel Mutations Affirm Their Importance as a Cause of Mitochondrial Disease. Hum. Mutat. 2013, 34, 1260–1268. [Google Scholar] [CrossRef]
- Yarham, J.W.; Elson, J.L.; Blakely, E.L.; McFarland, R.; Taylor, R.W. Mitochondrial tRNA mutations and disease. WIRES RNA 2010, 1, 304–324. [Google Scholar] [CrossRef]
- Pang, Y.L.J.; Poruri, K.; Martinis, S.A. tRNA synthetase: tRNA aminoacylation and beyond. WIRES RNA 2014, 5, 461–480. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Fu, R.; Wang, J.; Xiong, C.; Fan, Z.; Lu, D. Characterization of the mitochondrial genome of the pathogenic fungus Scytalidium auriculariicola (Leotiomycetes) and insights into its phylogenetics. Sci. Rep. 2019, 9, 17447. [Google Scholar] [CrossRef]
- Ma, Q.; Geng, Y.; Li, Q.; Cheng, C.; Zang, R.; Guo, Y.; Zhang, M. Comparative mitochondrial genome analyses reveal conserved gene arrangement but massive expansion/contraction in two closely related Exserohilum pathogens. Comput. Struct. Biotechnol. J. 2022, 20, 1456–1469. [Google Scholar] [CrossRef]
- Mamut, R.; FANG, J.J.; Anwar, G. Characterization and phylogenetic analysis of Ramalina sinensis mitogenome. Mycosystema 2023, 42, 1273–1284. [Google Scholar] [CrossRef]
- FANG, J.J.; Payzulla, T.; Anwar, G.; Mamut, R. Mitochondrial genome characteristics and phylogeny of Usnea lapponica. Genom. Appl. Biol. 2023, 42, 73–83. [Google Scholar] [CrossRef]
- Li, Q.; Yang, M.; Chen, C.; Xiong, C.; Jin, X.; Pu, Z.; Huang, W. Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus. Sci. Rep. 2018, 8, 9104. [Google Scholar] [CrossRef]
- Li, Q.; Ren, Y.; Shi, X.; Peng, L.; Zhao, J.; Song, Y.; Zhao, G. Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum Agaricomycotina. Int. J. Mol. Sci. 2019, 20, 5167. [Google Scholar] [CrossRef]
- Bibi, S.; Wang, D.; Wang, Y.; Mustafa, G.; Yu, H. Mitogenomic and Phylogenetic Analysis of the Entomopathogenic Fungus Ophiocordyceps lanpingensis and Comparative Analysis with Other Ophiocordyceps Species. Genes 2023, 14, 710. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, G.; Mamut, R.; Wang, J. Characterization of Complete Mitochondrial Genomes of the Five Peltigera and Comparative Analysis with Relative Species. J. Fungi 2023, 9, 969. https://doi.org/10.3390/jof9100969
Anwar G, Mamut R, Wang J. Characterization of Complete Mitochondrial Genomes of the Five Peltigera and Comparative Analysis with Relative Species. Journal of Fungi. 2023; 9(10):969. https://doi.org/10.3390/jof9100969
Chicago/Turabian StyleAnwar, Gulmira, Reyim Mamut, and Jiaqi Wang. 2023. "Characterization of Complete Mitochondrial Genomes of the Five Peltigera and Comparative Analysis with Relative Species" Journal of Fungi 9, no. 10: 969. https://doi.org/10.3390/jof9100969
APA StyleAnwar, G., Mamut, R., & Wang, J. (2023). Characterization of Complete Mitochondrial Genomes of the Five Peltigera and Comparative Analysis with Relative Species. Journal of Fungi, 9(10), 969. https://doi.org/10.3390/jof9100969