
The Identification and Comparative Analysis of Non-Coding RNAs in Spores and Mycelia of Penicillium expansum

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pathogen and Fungal Phenotype
2.2. Sample Preparation and Quantitative Validation
2.3. LncRNA-Sequencing Analysis
2.4. Small RNA-Sequencing Analysis
2.5. CircRNA-Sequencing Analysis
2.6. Statistical Analysis
3. Results
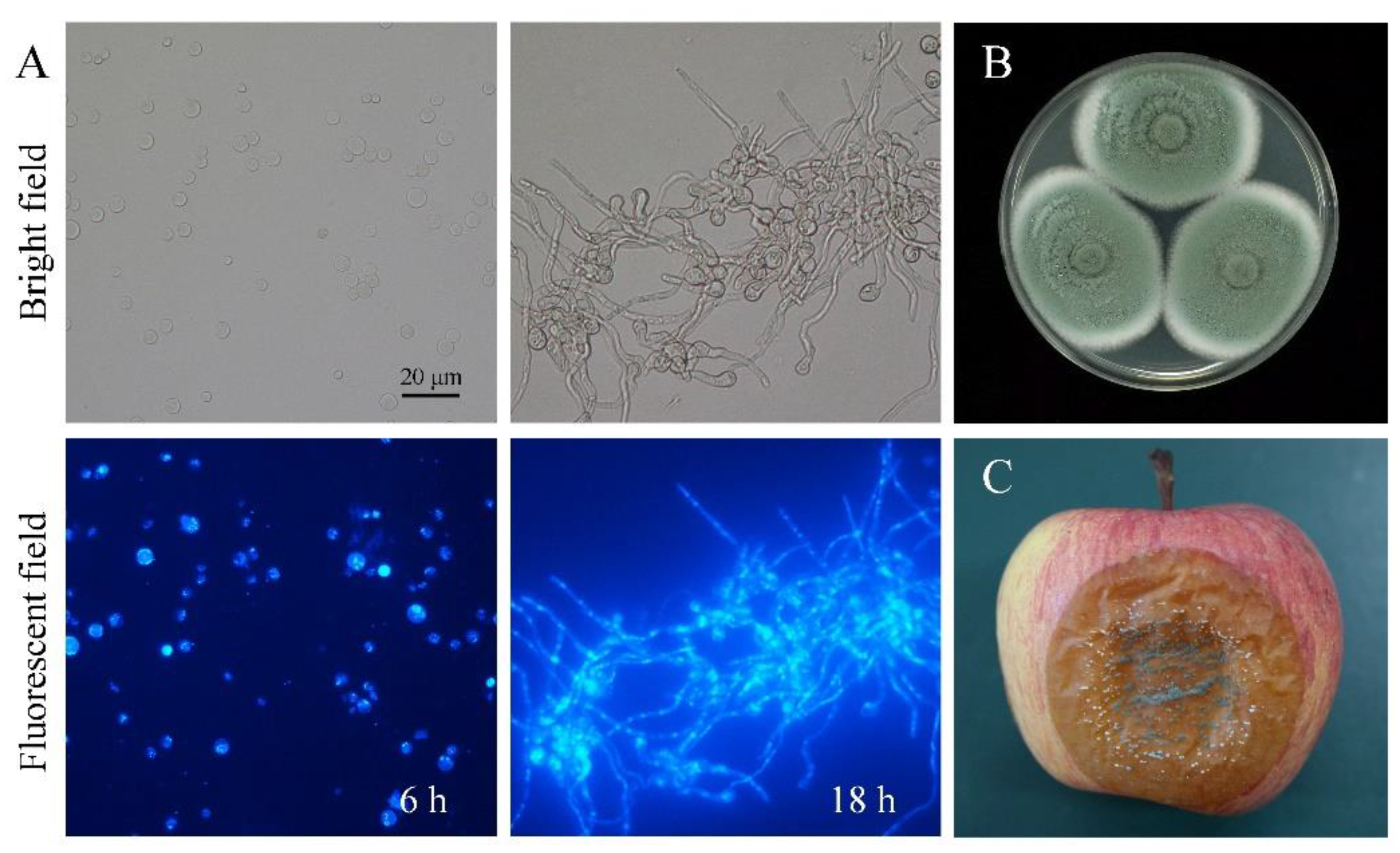
3.1. The Phenotype of P. expansum at Different Developmental Stages
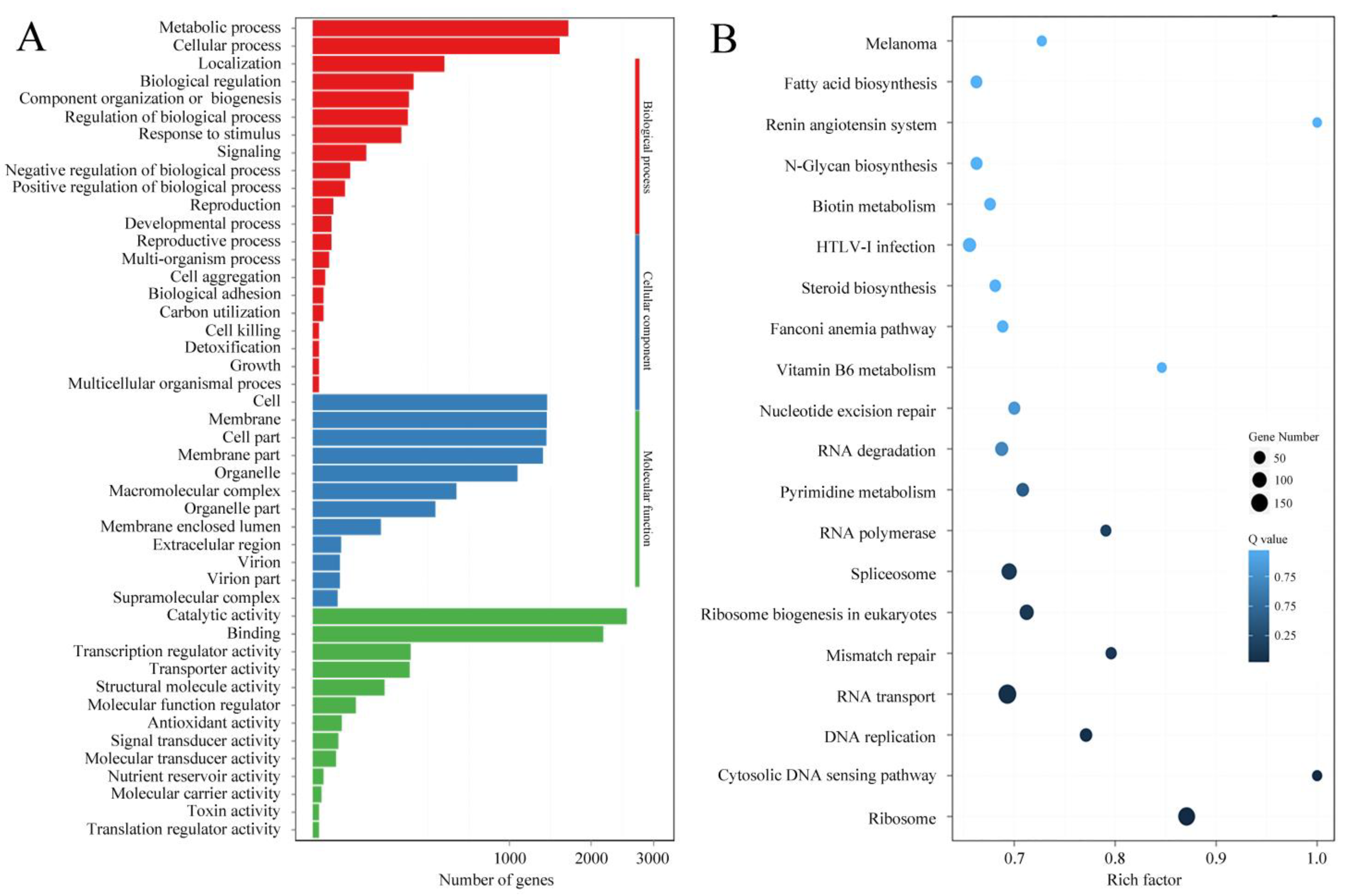
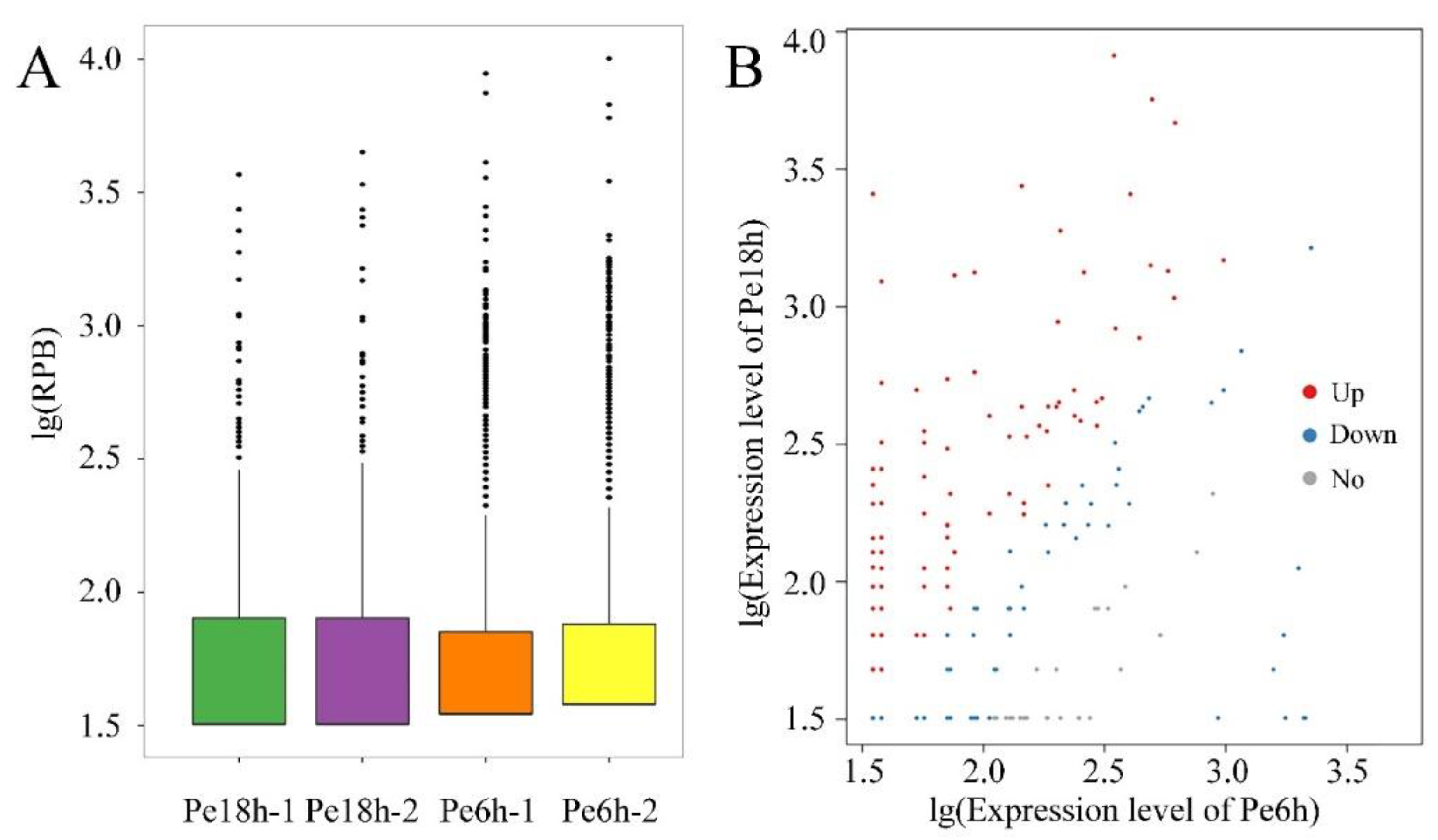
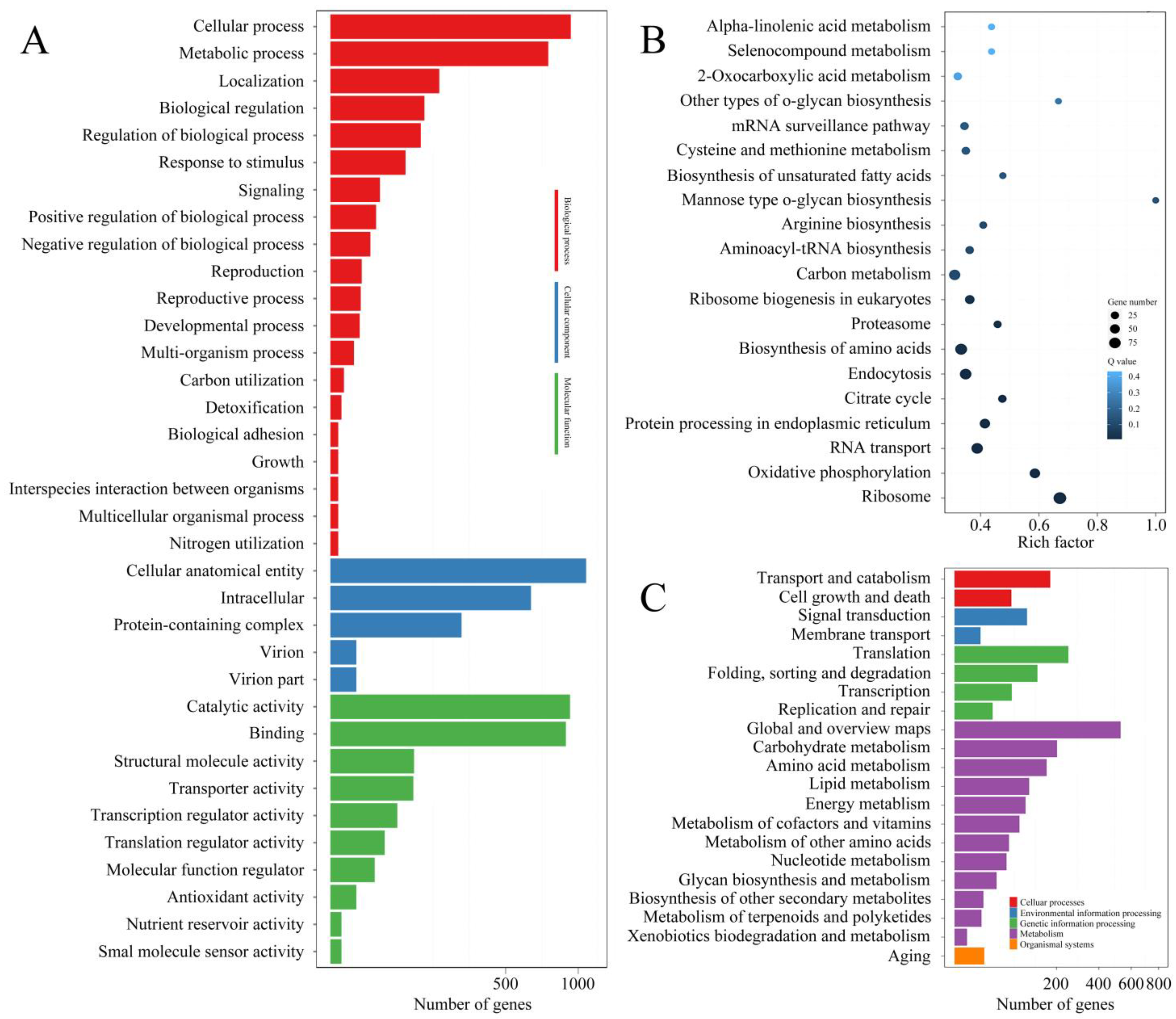
3.2. LncRNA-Sequencing Results
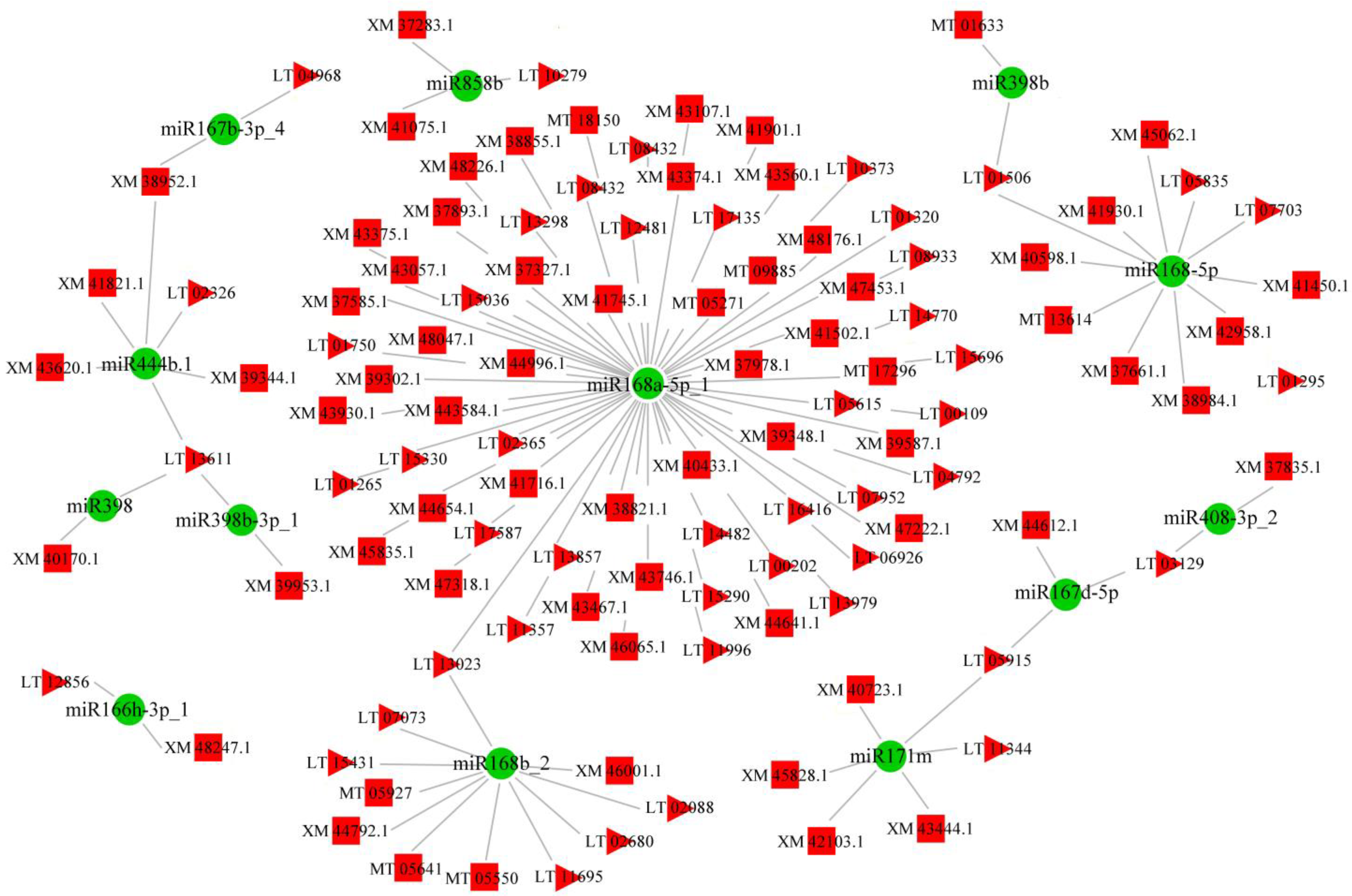
3.3. Small RNA-Sequencing Results
3.4. CircRNA-Sequencing Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, B.Q.; Chen, Y.; Zong, Y.Y.; Shang, Y.J.; Zhang, Z.Q.; Xu, X.D.; Wang, X.; Long, M.Y.; Tian, S.P. Dissection of patulin biosynthesis, spatial control and regulation mechanism in Penicillium expansum. Environ. Microbiol. 2019, 21, 1124–1139. [Google Scholar] [CrossRef]
- Li, C.Z.; Zhang, F.M.; Gan, D.; Wang, C.Y.; Zhou, H.; Yin, T.P.; Cai, L. Secondary metabolites isolated from Penicillium expansum and their chemotaxonomic value. Biochem. Syst. Ecol. 2023, 107, 104584. [Google Scholar] [CrossRef]
- Riachy, R.A.; Strub, C.; Durand, N.; Guibert, B.; Guichard, H.; Constancias, F.; Chochois, V.; Lopez-Lauri, F.; Fontana, A.; Schorr-Galindo, S. Microbiome status of cider-apples, from orchard to processing, with a special focus on Penicillium expansum occurrence and patulin contamination. J. Fungi 2021, 7, 244. [Google Scholar] [CrossRef]
- Maldonado, M.L.; Patriarca, A.; Cargo, P.M.; Lannone, L.; Sanchis, V.; Nielsen, K.F.; Pinto, V.F. Diversity and metabolomics characterization of Penicillium expansum isolated from apples grown in Argentina and Spain. Fungal Biol. 2022, 126, 547–555. [Google Scholar] [CrossRef]
- Yu, L.L.; Qiao, N.Z.; Zhao, J.X.; Zhang, H.; Tian, F.W.; Zhai, Q.X.; Chen, W. Postharvest control of Penicillium expansum in fruits: A review. Food Biosci. 2020, 36, 100633. [Google Scholar]
- Jimdjio, C.K.; Xue, H.L.; Bi, Y.; Nan, M.N.; Li, L.; Zhang, R.; Liu, Q.L.; Pu, L. Effect of ambient pH on growth, pathogenicity, and patulin production of Penicillium expansum. Toxins 2021, 13, 550. [Google Scholar] [CrossRef]
- Gong, D.; Bi, Y.; Zong, Y.Y.; Li, Y.C.; Sionov, E.; Prusky, D. Dynamic change of carbon and nitrogen sources in colonized apples by Penicillium expansum. Foods 2022, 11, 3367. [Google Scholar] [CrossRef]
- Pang, J.L.; Zhang, F.; Wang, Z.R.; Wu, Q.F.; Liu, B.J.; Meng, X.H. Inhibitory effect and mechanism of curcumin-based photodynamic inactivation on patulin secretion by Penicillium expansum. Innov. Food Sci. Emerg. 2022, 80, 103078. [Google Scholar] [CrossRef]
- Wang, M.; Du, Y.M.; Jiao, W.X.; Fu, M.R. Effects of fruit tissue pH value on the Penicillium expansum growth, patulin accumulation and distribution. J. Food Process. Preserv. 2022, 46, e168881. [Google Scholar] [CrossRef]
- Nicolau-Lapeña, I.; Rodríguez-Bencomo, J.J.; Colás-Medà, P.; Viñas, I.; Sanchis, V.; Alegre, I. Ultraviolet applications to control patulin produced by Penicillium expansum CMP-1 in apple products and study of further patulin degradation products formation and toxicity. Food Bioprocess Tech. 2023, 16, 804–823. [Google Scholar]
- Luciano-Rosario, D.; Keller, N.P.; Jurick, W.M., II. Penicillium expansum: Biology, omics, and management tools for a global postharvest pathogen causing blue mould of pome fruit. Mol. Plant Pathol. 2020, 21, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.; Ngea, G.L.N.; Godana, E.A.; Shi, Y.; Lanhuang, B.; Zhang, X.Y.; Zhao, L.N.; Yang, Q.Y.; Wang, S.Y.; Zhang, H.Y. Recent advances in Penicillium expansum infection mechanism and current methods in controlling P. expansum in postharvest apples. Crit. Rev. Food Sci. 2023, 63, 2598–3611. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Yang, Q.Y.; Boateng, N.A.S.; Ahima, J.; Dou, Y.; Zhang, H.Y. Ultrastructure observation and transcriptome analysis of Penicillium expansum invasion in postharvest pears. Postharvest Biol. Technol. 2020, 165, 111198. [Google Scholar]
- Wang, Y.R.; Yang, Q.Y.; Godana, E.A.; Zhang, Y.; Zhang, H.Y. Ultrastructural observation and transcriptome analysis provide insights into mechanisms of Penicillium expansum invading apple wounds. Food Chem. 2023, 414, 135633. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Shu, Y.L.; Quan, S.H.; Dhanasekaran, S.; Zhang, X.Y.; Zhang, H.Y. Screening and regulation mechanism of key transcription factors of Penicillium expansum infecting postharvest pears by ATAC-Seq analysis. Foods 2022, 11, 3855. [Google Scholar] [CrossRef] [PubMed]
- Tannous, J.; Barda, O.; Luciano-Rosario, D.; Prusky, D.B.; Sionov, E.; Keller, N.P. New insight into pathogenicity and secondary metabolism of the plant pathogen Penicillium expansum through deletion of the epigenetic reader SntB. Front. Microbiol. 2020, 11, 610. [Google Scholar]
- Zhang, X.M.; Zong, Y.Y.; Gong, D.; Yu, L.R.; Sionov, E.; Bi, Y.; Prusky, D. NADPH oxidase regulates the growth and pathogenicity of Penicillium expansum. Front. Plant Sci. 2021, 12, 696210. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Meng, D.; Xia, X.S.; Sun, Y.M.; Zhao, L.N.; Zhou, X.H. Profiling the secretomes of Penicillium expansum reveals that a serine carboxypeptidase (PeSCP) is required for the fungal virulence on apple fruit. Physiol. Mol. Plant Pathol. 2022, 122, 101897. [Google Scholar] [CrossRef]
- Zhang, X.M.; Zong, Y.Y.; Gong, D.; Zhang, F.; Yu, L.R.; Bi, Y.; Sionov, E.; Prusky, D. Small GTPase RacA is critical for spore growth, patulin accumulation, and virulence of Penicillium expansum. Postharvest Biol. Tec. 2022, 191, 111964. [Google Scholar] [CrossRef]
- Xu, X.D.; Chen, Y.; Li, B.Q.; Tian, S.P. Arginine methylatransferase PeRmtC regulates development and pathogenicity of Penicillium expansum via mediating key genes in conidiation and secondary metabolism. J. Fungi 2021, 7, 807. [Google Scholar] [CrossRef]
- Xu, M.Q.; Zhang, Q.D.; Dhanasekaran, S.; Godana, E.A.; Zhang, X.Y.; Yang, Q.Y.; Zhao, L.N.; Zhang, H.Y. The necrosis-inducing protein (NIP) gene contributes to Penicillium expansum virulence during postharvest pear infection. Food Res. Int. 2022, 158, 111562. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Chen, Y.; Li, B.Q.; Tian, S.P. Histone H3K4 methyltransferase PeSet1 regulates colonization, patulin biosynthesis, and stress responses of Penicillium expansum. Microbiol. Spectr. 2023, 11, 1. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Gong, W.F.; Tu, T.T.; Zhang, J.Q.; Xia, X.S.; Zhao, L.N.; Zhou, X.H.; Wang, Y. Transcriptome analysis and functional characterization reveal that Peclg gene contributes to the virulence of Penicillium expansum on apple fruits. Foods 2023, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Tannous, J.; Khoury, R.E.; Snini, S.P.; Lippi, Y.; Khoury, A.E.; Atoui, A.; Lteif, R.; Oswald, I.P.; Puel, O. Sequencing, physical organization and kinetic expression of the patulin biosynthetic gene cluster from Penicillium expansum. Int. J. Food Microbiol. 2014, 189, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Tannous, J.; Keller, N.P.; Atoui, A.; Khoury, A.E.; Lteif, R.; Oswald, I.P.; Puel, O. Secondary metabolism in Penicillium expansum: Emphasis on recent advances in patulin research. Crit. Rev. Food Sci. 2017, 58, 2082–2098. [Google Scholar] [CrossRef]
- Desgranges, E.; Marzi, S.; Moreau, K.; Romby, P.; Caldelari, I. Noncoding RNA. Microbiol. Spectr. 2019, 7, GPP3-0038-2018. [Google Scholar]
- Qian, X.Y.; Zhao, J.Y.; Yeung, P.Y.; Zhang, Q.F.; Kwok, C.K. Revealing lncRNA structures and interactions by sequencing-based approaches. Trends Biochem. Sci. 2019, 44, 33–53. [Google Scholar] [CrossRef]
- Bridges, M.C.; Daulagala, A.C.; Kourtidis, A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021, 220, e202009045. [Google Scholar]
- Jinek, M.; Doudan, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [PubMed]
- Zhang, P.; Dai, M.Q. CircRNA: A rising star in plant biology. J. Genet. Genom. 2022, 49, 1081–1092. [Google Scholar] [CrossRef]
- Islam, W.; Islam, S.U.; Qasim, M.; Wang, L. Host-Pathogen interactions modulated by small RNAs. RNA Biol. 2017, 14, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Avrova, A.Q.; Whisson, S.C.; Prithard, L.; Venter, E. A novel non-protein-coding infection-specific gene family is clustered throughout the genome of Phytophthora infestans. Microbiology 2007, 153, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Till, P.; Pucher, M.E.; Mach, R.L.; Mach-Aigner, A.R. A long noncoding RNA promotes cellulose expression in Trichoderma reesei. Biotechnol. Biofuels 2018, 11, 78. [Google Scholar] [CrossRef]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.W.; Zhang, Z.H.; Kaloshian, I.; Huang, H.D.; Jin, H.L. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathway. Science 2013, 342, 118–123. [Google Scholar] [CrossRef]
- Fahlgren, N.; Bollmann, S.R.; Kasschau, K.D.; Cuperus, J.T.; Press, C.M.; Sullivan, C.M.; Chapman, E.J.; Hoyer, J.S.; Gilbert, K.B.; Grünwald, N.J.; et al. Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS ONE 2013, 8, e77181. [Google Scholar]
- Lai, T.F.; Wang, Y.; Fan, Y.Y.; Zhou, Y.Y.; Bao, Y.; Zhou, T. The response of growth and patulin production of postharvest pathogen Penicillium expansum to exogenous potassium phoshite treatment. Int. J. Food Microbiol. 2017, 244, 1–10. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Komienko, A.E.; Guenzl, P.M.; Barlow, D.P.; Pauler, F.M. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013, 11, 59. [Google Scholar]
- Friedländer, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.; Hutter, M.; Dueck, A.; Meister, G.; Engelmann, J.C. miRA: Adaptable novel miRNA identification in plants using small RNA sequencing data. BMC Bioinform. 2015, 15, 370. [Google Scholar] [CrossRef] [PubMed]
- Jagla, B.; Aulner, N.; Kelly, P.D.; Song, D.; Volchuk, A.; Zatorski, A.; Shum, D.; Mayer, T.; De Angelis, D.A.; Ouerfelli, O.; et al. Sequence characteristics of functional siRNAs. RNA 2005, 11, 864–872. [Google Scholar] [CrossRef]
- Bonnet, E.; He, Y.; Billiau, K.; De Peer, Y.V. TAPIR, a web server for the prediction of plant microRNA targets including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, 22–28. [Google Scholar] [CrossRef]
- Memcazk, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.F.; Zhao, F.Q. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Ballester, A.; Marcet-Houben, M.; Levin, E.; Sela, N.; Selma-Lázaro, C.; Carmona, L.; Wisniewski, M.; Droby, S.; González-Candelas, L.; Gabaldón, T. Genome, transcriptome and functional analyses of Penicillium expansum provide new insights into secondary metabolism and pathogenicity. Int. J. Food Microbiol. 2015, 28, 232–248. [Google Scholar] [CrossRef]
- Li, B.; Zong, Y.Y.; Du, Z.L.; Chen, Y.; Zhang, Z.Q.; Qin, G.Z.; Zhao, W.M.; Tian, S.P. Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in Penicillium species. Mol. Plant Microbe Interact. 2015, 28, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, X.H.; Luo, J.; Ye, B.S.; Zhou, Y.Y.; Zhou, L.W.; Lai, T.F. Identification of differentially expressed genes involved in spore germination of Penicillium expansum by comparative transcriptome and proteome approaches. Microbiologyopen 2018, 7, e562. [Google Scholar] [CrossRef] [PubMed]
- Chacko, N.; Lin, X.R. Non-coding RNAs in the development and pathogenesis of eukaryotic microbes. Appl. Microbiol. Biotechnol. 2013, 97, 7989–7997. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.F.; Bian, Q.; He, X.W.; Zhang, H.; He, J.J. A lncRNA-miRNA-mRNA ceRNA regulatory network based tuberculosis prediction model. Microb. Pathog. 2021, 158, 105069. [Google Scholar] [CrossRef]
- Tan, J.J.; Li, X.Y.; Zhang, L.; Du, Z.L. Recent advances in machine learning methods for predicting LncRNA and disease associations. Front. Cell. Infect. Microbiol. 2022, 12, 1071972. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.X.; Chow, E.W.L.; Wang, H.T.; Xu, X.L.; Cai, C.L.; Song, Y.B.; Wang, J.B.; Wang, Y. LncRNA DINOR is a virulence factor and global regulator of stress responses in Candida auris. Nat. Microbiol. 2021, 6, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Menegidio, F.B.; Barbosa, D.A.; Alencar, V.C.; Boas, R.O.V.; De Oliveira, R.C.; Jabes, D.L.; Nunes, L.R. Transcriptomic profiling identifies novel transcripts, isomorphs, and noncoding RNAs in Paracoccidioides brasiliensis. Med. Mycol. 2021, 59, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.; Saville, B.J. Ustilago maydis natural antisense transcript expression alters mRNA stability and pathogenesis. Mol. Microbiol. 2013, 89, 29–51. [Google Scholar] [CrossRef]
- Arthanari, Y.; Heintzen, C.; Griffiths-Jones, S.; Crosthwaite, S.K. Natural antisense transcripts and long non-coding RNA in Neurospora crassa. PLoS ONE 2014, 9, e91353. [Google Scholar]
- Yang, H.; Wang, X.F.; Li, Z.J.; Guo, Q.B.; Yang, M.G.; Chen, D.; Wang, C.L. The effect of blue light on the production of citrinin in Monascus purpureus M9 by regulating the mraox gene through lncRNA AOANCR. Toxins 2019, 11, 536. [Google Scholar] [CrossRef]
- Neumeier, J.; Meister, G. siRNA specificity: RNAi mechanisms and strategies to reduce off-target effects. Front. Plant Sci. 2021, 11, 526455. [Google Scholar] [PubMed]
- Yadav, A.; Sanyal, I.; Rai, S.P.; Lata, C. An overview on miRNA-encoded peptides in plant biology research. Genomics 2021, 113, 2385–2391. [Google Scholar] [CrossRef]
- Achkar, N.P.; Cambiagno, D.A.; Manavella, P.A. miRNA biogenesis: A dynamic pathway. Trends Plant Sci. 2016, 21, 1034–1044. [Google Scholar] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [PubMed]
- Lee, H.C.; Li, L.; Gu, W.F.; Xue, Z.H.; Crosthwaite, S.K.; Pertsemlidis, A.; Lewis, Z.A.; Freitag, M.; Selker, E.U.; Mello, C.C.; et al. Diverse pathways generate microRNA-like RNAs and dicer-independent small interfering RNAs in fungi. Mol. Cell 2010, 38, 803–814. [Google Scholar] [CrossRef]
- Lin, Y.L.; Ma, L.T.; Lee, Y.R.; Lin, S.S.; Wang, S.Y.; Chang, T.T.; Shaw, J.F.; Li, W.H.; Chu, F.H. MicroRNA-like small RNAs prediction in the development of Antrodia cinnamonmea. PLoS ONE 2015, 10, e0123245. [Google Scholar]
- Wang, B.; Sun, Y.F.; Song, N.; Zhao, M.X.; Liu, R.; Feng, H.; Wang, X.J.; Kang, Z.S. Puccinia striiformis f. sp. tritici microRNA-like RNA 1 (Pst-milR1), an important pathogenicity factor of Pst, impairs wheat resistance to Pst by suppressing the wheat pathogenesis-related 2 gen. New Phytol. 2017, 215, 338–350. [Google Scholar] [CrossRef]
- Jin, Y.; Zhao, J.H.; Zhao, P.; Zhang, T.; Wang, S.; Guo, H.S. A fungal milRNA mediates epigenetic repression of a virulence gene in Verticillium dahlia. Philos. Trans. R. Soc. B 2019, 374, 20180309. [Google Scholar] [CrossRef]
- Hu, X.Y.; Hodén, K.P.; Liao, Z.; Åsman, A.; Dixelius, C. Phytophthora infestans Ago1-associated miRNA promotes potato late blight disease. New Phytol. 2022, 233, 443–457. [Google Scholar] [CrossRef]
- Khatri, M.; Rajam, M.V. Targeting polyamines of Aspergillus nidulans by siRNA specific to fungal ornithine decarboxylase gene. Med. Mycol. 2007, 45, 211–220. [Google Scholar] [CrossRef]
- Hammond, T.M.; Xiao, H.; Boone, E.C.; Decker, L.M.; Lee, S.A.; Perdue, T.D.; Pukkila, P.J.; Shiu, P.K.T. Novel proteins required for meiotic silencing by unpaired DNA and siRNA generation in Neurospora crassa. Genetics 2013, 194, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, P.Y.; Feng, H.W.; Zhao, Q.; Liu, H.S. Using network distance analysis to predict lncRNA-miRNA interactions. Interdiscip. Sci. 2021, 13, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Gazzani, S.; Li, M.; Maistri, S.; Scarponi, E.; Graziola, M.; Barbaro, E.; Wunder, J.; Furini, A.; Saedler, H.; Varotto, C. Evolution of MIR168 paralogs in Barassicaceae. BMC Evol. Biol. 2009, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Vaucheret, H. AGO1 homeostasis involves differential production of 21-nt and 22-nt miR168 species by MIR168a and MIR168b. PLoS ONE 2009, 4, e6442. [Google Scholar] [CrossRef] [PubMed]
- Iki, T.; Cléry, A.; Bologna, N.G.; Sarazin, A.; Brosnan, C.A.; Pumplin, N.; Allain, F.H.T.; Voinnet, O. Structural flexibility enables alternative maturation, ARGONAUTE sorting and activities of miR168, a global gene silencing regulator in plants. Mol. Plant 2018, 11, 1008–1023. [Google Scholar] [CrossRef]
- Vaucheret, H.; Mallory, A.C.; Bartel, D.P. AGO1 homeostasis entails coexpression of MIR168 and AGO1 and preferential stabilization of miR168 by AGO1. Mol. Cell 2006, 22, 129–139. [Google Scholar] [CrossRef]
- Dalmadi, Á.; Miloro, F.; Bálint, J.; Várallyay, É.; Havelda, Z. Controlled RISC loading efficiency of miR168 defined by miRNA duplex structure adjusts ARGONAUTE1 homeostasis. Nucleic Acids Res. 2021, 49, 12912–12928. [Google Scholar] [CrossRef]
- Baldrich, P.; Kakar, K.; Siré, C.; Moreno, A.B.; Berger, A.; Garcia-Chapa, M.; López-Moya, J.J.; Riechmann, J.L.; Segundo, B.S. Small RNA profiling reveals regulation of Arabidopsis miR168 and heterochromatic siRNA415 in response to fungal elicitors. BMC Genom. 2014, 15, 1083. [Google Scholar]
- Xian, Z.Q.; Huang, W.; Yang, Y.W.; Tang, N.; Zhang, C.; Ren, M.Z.; Li, Z.G. miR168 influences phase transition, leaf epinasty, and fruit development via SlAGO1s in tomato. J. Exp. Bot. 2014, 65, 6655–6666. [Google Scholar] [CrossRef]
- Yu, X.Y.; Hou, Y.J.; Chen, W.P.; Wang, S.H.; Wang, P.H.; Qu, S.C. Malus hupehensis miR168 targets to ARGONAUTE1 and contributes to the resistance against Botryosphaeria dothidea infection by altering defense responses. Plant Cell Physiol. 2017, 58, 1541–1557. [Google Scholar]
- Liu, X.; Tan, C.C.; Cheng, X.; Zhao, X.M.; Li, T.L.; Jiang, J. miR168 targets Argonaute1A mediated miRNAs regulation pathways in response to potassium deficiency stress in tomato. BMC Plant Biol. 2020, 20, 477. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Chern, M.; Zhu, Y.; Zhang, L.L.; Lu, J.H.; Li, X.P.; Dang, W.Q.; Ma, X.C.; Yang, Z.R.; et al. Suppression of rice miR168 improves yield, flowering timer and immunity. Nat. Plants 2021, 7, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, D.X.; Chen, X.; Li, D.H.; Zhu, L.Y.; Zhang, Y.J.; Zhang, Y.J.; Li, J.; Bian, Z.; Liang, X.Y.; et al. Exogenous plant MIR168a specifically targets mammalian LDLPAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2012, 22, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Kuranaga, Y.; Heishima, K.; Sugito, N.; Morikawa, K.; Ito, Y.; Soga, T.; Ito, T. Plant hvu-MIR158-3p enhances expression of glucose transporter 1 (SLC2A1) in human cells by silencing genes related to mitochondrial electron transport chain complex I. J. Nutr. Biochem. 2022, 101, 108922. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, N.N.; Yang, L.; Tang, J.J.; Wang, Y.L.; Mei, M. A brief review of circRNA biogenesis, detection, and function. Curr. Genom. 2021, 22, 485–495. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, T.; Yu, Q.; Pan, J.; Wang, J.; Tang, Z.; Bai, X.; Shi, L.; Zhou, T. The Identification and Comparative Analysis of Non-Coding RNAs in Spores and Mycelia of Penicillium expansum. J. Fungi 2023, 9, 999. https://doi.org/10.3390/jof9100999
Lai T, Yu Q, Pan J, Wang J, Tang Z, Bai X, Shi L, Zhou T. The Identification and Comparative Analysis of Non-Coding RNAs in Spores and Mycelia of Penicillium expansum. Journal of Fungi. 2023; 9(10):999. https://doi.org/10.3390/jof9100999
Chicago/Turabian StyleLai, Tongfei, Qinru Yu, Jingjing Pan, Jingjing Wang, Zhenxing Tang, Xuelian Bai, Lue Shi, and Ting Zhou. 2023. "The Identification and Comparative Analysis of Non-Coding RNAs in Spores and Mycelia of Penicillium expansum" Journal of Fungi 9, no. 10: 999. https://doi.org/10.3390/jof9100999
APA StyleLai, T., Yu, Q., Pan, J., Wang, J., Tang, Z., Bai, X., Shi, L., & Zhou, T. (2023). The Identification and Comparative Analysis of Non-Coding RNAs in Spores and Mycelia of Penicillium expansum. Journal of Fungi, 9(10), 999. https://doi.org/10.3390/jof9100999