
An Efficient CRISPR/Cas9 Genome Editing System for a Ganoderma lucidum Cultivated Strain by Ribonucleoprotein Method
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Preparation of Protoplasts
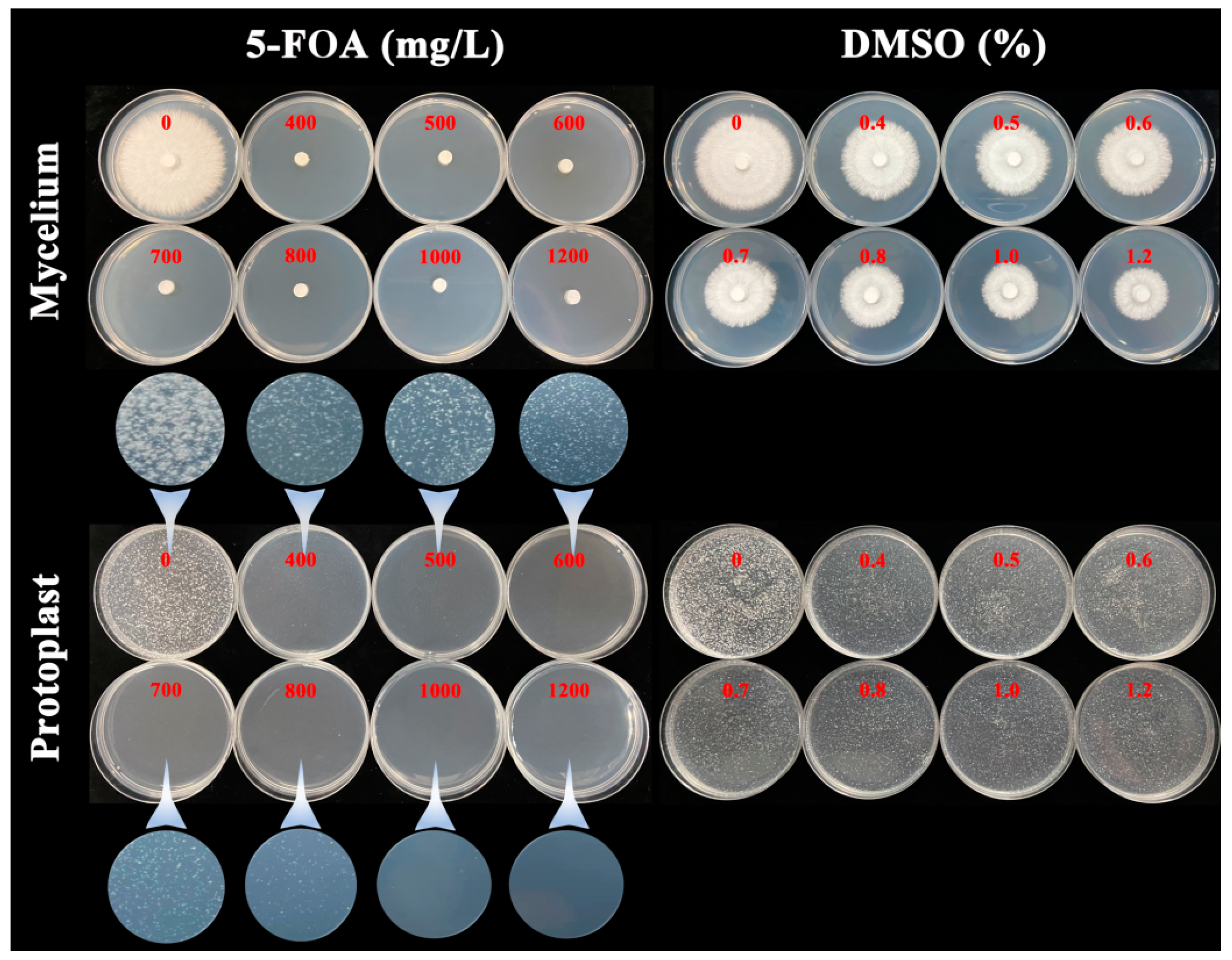
2.2. The 5-FOA Lethal Experiment on G. lucidum Strain L1
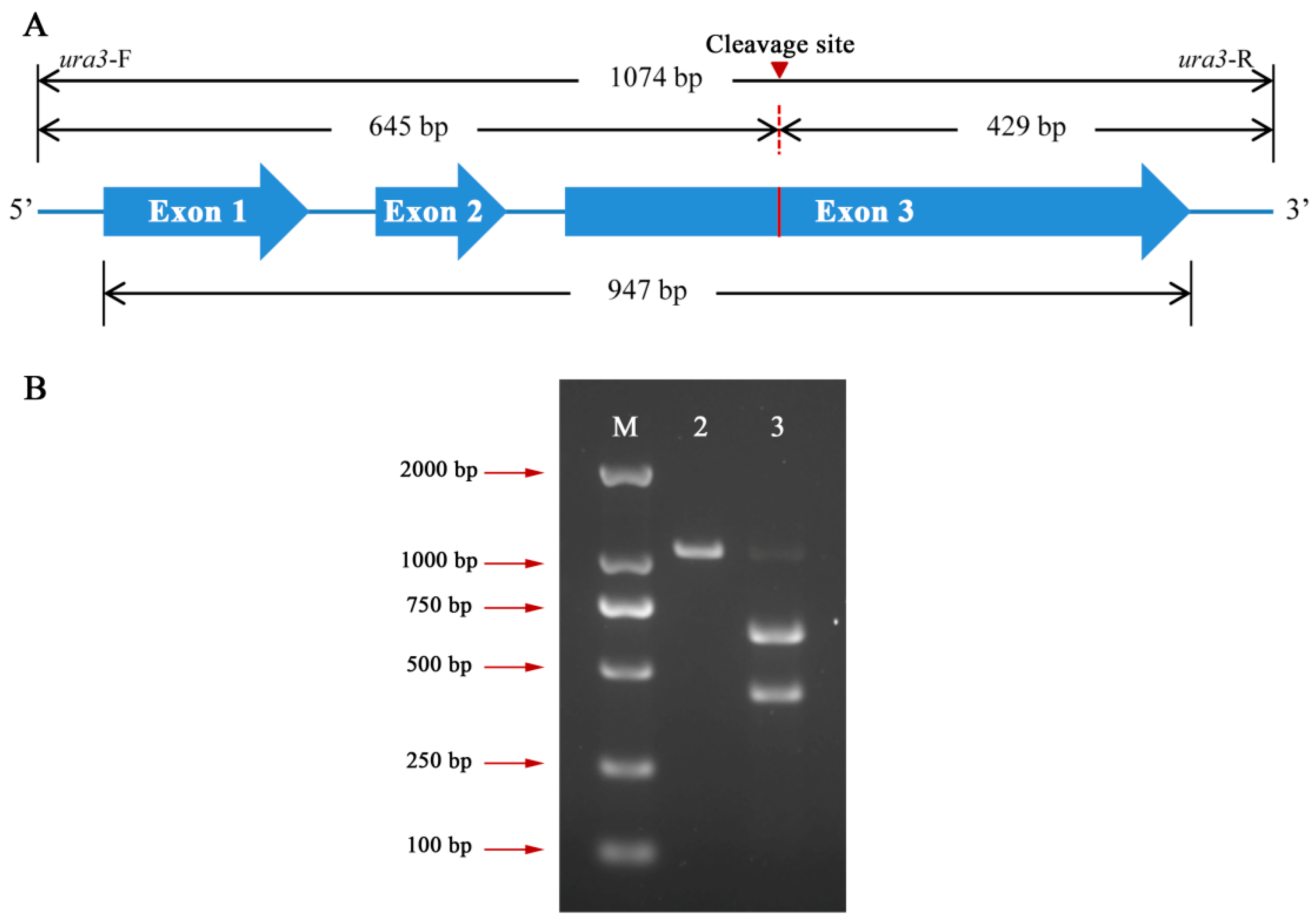
2.3. Sequencing of ura3, cyp512a3, and cyp5359n1 from G. lucidum Strain L1
2.4. Preparation of sgRNA-ura3 and Cas9 Protein
2.5. In Vitro Cas9 Cleavage Assay
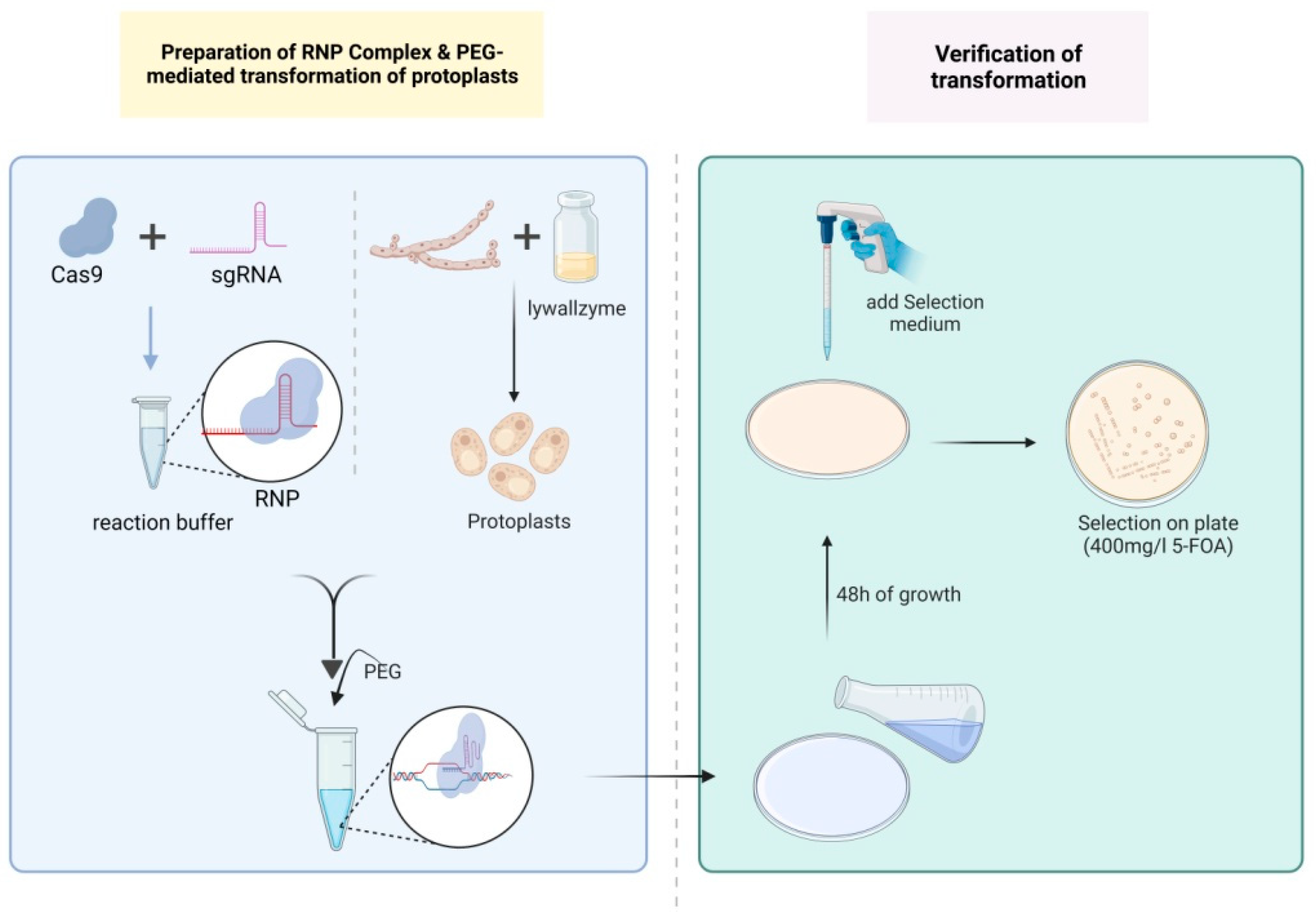
2.6. PEG-Mediated Transformation of Protoplasts
2.7. Screening and Verification of Transformants
3. Results
3.1. The 5-FOA Lethal Experiment on G. lucidum Strain L1
3.2. In Vitro Cas9 Cleavage Assay
3.3. Optimization of PEG-Mediated Protoplast Transformation of RNPs on G. lucidum Strain L1
3.4. Analysis of the Repair Characteristics of DSB Induced by RNA-Programmed Nuclease Cas9
3.5. Editing of Functional Genes in G. lucidum Using the Constructed CRISPR/Cas9 System
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanodiya, B.S.; Thakur, G.S.; Baghel, R.K.; Prasad, G.B.; Bisen, P.S. Ganoderma lucidum: A Potent Pharmacological Macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Zhang, S.Y.; Peng, B.; Tan, D.C.; Wu, M.Y.; Wei, J.C.; Wang, Y.T.; Luo, H. Ganoderma lucidum: A Comprehensive Review of Phytochemistry, Efficacy, Safety and Clinical Study. Food Sci. Hum. Wellness 2024, 13, 568–596. [Google Scholar] [CrossRef]
- Li, J.Q.; Zhang, J.H.; Chen, H.M.; Chen, X.D.; Lan, J.; Liu, C. Complete Mitochondrial Genome of the Medicinal Mushroom Ganoderma lucidum. PLoS ONE 2013, 8, e72038. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Xu, J.; Liu, C.; Zhu, Y.J.; Nelson, D.R.; Zhou, S.G.; Li, C.F.; Wang, L.Z.; Guo, X.; Sun, Y.Z.; et al. Genome Sequence of the Model Medicinal Mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Xiao, H.; Zou, G.; Zhou, Z.H.; Zhong, J.J. CRISPR-Cas9 Assisted Gene Disruption in the Higher Fungus Ganoderma Species. Process Biochem. 2017, 56, 57–61. [Google Scholar] [CrossRef]
- Li, H.H.; Liu, G. The Application of CRISPR/Cas9 in Genome Editing of Filamentous Fungi. Hereditas 2017, 39, 355–367. [Google Scholar] [PubMed]
- Bari, V.K.; Nassar, J.A.; Aly, R. CRISPR/Cas9 Mediated Mutagenesis of MORE AXILLARY GROWTH 1 in Tomato Confers Resistance to Root Parasitic Weed Phelipanche aegyptiaca. Sci. Rep. 2021, 11, 3905. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Futamata, R.; Horibe, T.; Ueda, K.; Kinoshita, M. CRISPR/Cas9 Nickase-Mediated Efficient and Seamless Knock-in of Lethal Genes in the Medaka Fish Oryzias latipes. Dev. Growth Differ. 2020, 62, 554–567. [Google Scholar] [CrossRef]
- Schuster, M.; Kahmann, R. CRISPR-Cas9 Genome Editing Approaches in Filamentous Fungi and Oomycetes. Fungal Genet. Biol. 2019, 130, 43–53. [Google Scholar] [CrossRef]
- Liu, K.; Sun, B.; You, H.; Tu, J.L.; Yu, X.Y.; Zhao, P.; Xu, J.W. Dual sgRNA-directed Gene Deletion in Basidiomycete Ganoderma lucidum Using the CRISPR/Cas9 System. Microbiol. Biotechnol. 2020, 13, 386–396. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhang, J.S.; Zou, G.; Tang, C.H.; Feng, J.; Bao, D.P.; Chen, J.B.; Tan, Y. Construction of a CRISPR/Cas9-Based Genome Editing System in Ganoderma lucidum ‘Hunong No.1’ Cultivar. Acta Edulis Fungi 2023, 30, 9–18. [Google Scholar]
- Wang, P.A.; Xiao, H.; Zhong, J.J. CRISPR-Cas9 Assisted Functional Gene Editing in the Mushroom Ganoderma lucidum. Appl. Microbiol. Biotechnol. 2020, 104, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.; Choi, Y.J.; Nandre, R.; Han, H.G.; Kim, S.; Kim, M.; Oh, Y.L.; Nakazawa, T.; Honda, Y.; Ro, H.S. The Cas9-gRNA Ribonucleoprotein Complex-Mediated Editing of PyrG in Ganoderma lucidum and Unexpected Insertion of Contaminated DNA Fragments. Sci. Rep. 2023, 13, 11133. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.S.; Shi, L.; Ren, A.; Li, M.J.; Wu, F.L.; Jiang, A.L.; Zhao, M.W. The Development and Application of a Multiple Gene Co-Silencing System Using Endogenous URA3 as a Reporter Gene in Ganoderma lucidum. PLoS ONE 2012, 7, e43737. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, Y.X. Progress and Challenge of the CRISPR-Cas System in Gene Editing for Filamentous Fungi. Synth. Biol. J. 2021, 2, 274–286. [Google Scholar]
- Enkler, L.; Richer, D.; Marchand, A.L.; Ferrandon, D.; Jossinet, F. Genome Engineering in the Yeast Pathogen Candida glabrata Using the CRISPR-Cas9 System. Sci. Rep. 2016, 6, 35766. [Google Scholar] [CrossRef]
- Cho, J.S.; Choi, K.R.; Prabowo, C.P.S.; Shin, J.H.; Yang, D.; Jang, J.; Lee, S.Y. CRISPR/Cas9-Coupled Recombineering for Metabolic Engineering of Corynebacterium glutamicum. Metab. Eng. 2017, 42, 157–167. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Jan Vonk, P.; Escobar, N.; Wösten, H.A.B.; Lugones, L.G.; Ohm, R.A. High-throughput targeted gene deletion in the model mushroom Schizophyllum commune using pre-assembled Cas9 ribonucleoproteins. Sci. Rep. 2019, 9, 7632. [Google Scholar] [CrossRef]
- Zou, G.; Xiao, M.L.; Chai, S.X.; Zhu, Z.H.; Wang, Y.; Zhou, Z.H. Efficient Genome Editing in Filamentous Fungi via an Improved CRISPR-Cas9 Ribonucleoprotein Method Facilitated by Chemical Reagents. Microbiol. Biotechnol. 2021, 14, 2343–2355. [Google Scholar] [CrossRef]
- Yu, L.; Xiao, M.L.; Zhu, Z.H.; Wang, Y.M.; Zhou, Z.H.; Wang, P.P.; Zou, G. Efficient genome editing in Claviceps purpurea using a CRISPR/Cas9 ribonucleoprotein method. Synth. Syst. Biotechnol. 2022, 7, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Ji, S.L.; He, Y.L.; Ren, M.F.; Xu, J.W. Development of an Expression Plasmid and Its Use in Genetic Manipulation of Lingzhi or Reishi Medicinal Mushroom, Ganoderma lucidum (Higher Basidiomycetes). Int. J. Med. Mushrooms 2014, 16, 161–168. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.T.; Shen, M.Y.; Liu, J.Y.; Li, Y.R.; Xu, S.; Chen, L.; Shi, G.Y.; Ding, Z.Y. Establishment of an Efficient Polyethylene Glycol (PEG)-Mediated Transformation System in Pleurotus eryngii var. ferulae Using Comprehensive Optimization and Multiple Endogenous Promoters. J. Fungi 2022, 8, 186. [Google Scholar]
- Kües, U.; Nelson, D.R.; Liu, C.; Yu, G.J.; Zhang, J.H.; Li, J.Q.; Wang, X.C.; Sun, H. Genome Analysis of Medicinal Ganoderma Spp. with Plant-Pathogenic and Saprotrophic Life-Styles. Phytochemistry 2015, 114, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Ahyayauch, H.; Collado, M.I.; Alonso, A.; Goñi, F.M. Lipid Bilayers in the Gel Phase Become Saturated by Triton X-100 at Lower Surfactant Concentrations Than Those in the Fluid Phase. Biophys. J. 2012, 102, 2510–2516. [Google Scholar] [CrossRef]
- Mattei, B.; Lira, R.B.; Perez, K.R.; Riske, K.A. Membrane Permeabilization Induced by Triton X-100: The Role of Membrane Phase State and Edge Tension. Chem. Phys. Lipids 2017, 202, 28–37. [Google Scholar] [CrossRef]
- Gao, W.; Baars, J.J.P.; Maliepaard, C.; Visser, R.G.F.; Zhang, J.X.; Sonnenberg, A.S.M. Multi-Trait QTL Analysis for Agronomic and Quality Characters of Agaricus bisporus (Button Mushrooms). AMB Express 2016, 6, 67. [Google Scholar] [CrossRef]
- Zuo, W.L.; Chao, Q.; Zhang, N.; Ye, J.R.; Tan, G.Q.; Li, B.L.; Xing, Y.X.; Zhang, B.Q.; Liu, H.J.; Fengler, K.A.; et al. A Maize Wall-Associated Kinase Confers Quantitative Resistance to Head Smut. Nat. Genet. 2015, 47, 151–157. [Google Scholar] [CrossRef]
- Chai, S.X.; Zhu, Z.H.; Tian, E.N.; Xiao, M.L.; Wang, Y.; Zou, G.; Zhou, Z.H. Building a Versatile Protein Production Platform Using Engineered Trichoderma reesei. ACS Synth. Biol. 2022, 11, 486–496. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNP (nM) | Colony-Forming Units (CFUs) | Deletion Rate | Insertion Rate | Proportion of Large Fragments among All Insertions | Substitution Rate |
|---|---|---|---|---|---|
| 0.0 | 0 | 0 | 0 | 0 | 0 |
| 73.5 | 39 (10/10) a | 30.0% (3/10) b | 50.0% (5/10) c | 20.0% (1/5) d | 20.0% (2/10) e |
| 147.0 | 83 (10/10) a | 70.0% (7/10) b | 30.0% (3/10) c | 66.7% (2/3) d | 0 |
| 220.6 | 100 (35/35) a | 40.0% (14/35) b | 48.6% (17/35) c | 76.5% (13/17) d | 11.4% (4/35) e |
| 294.0 | 111 (35/35) a | 57.1% (20/35) b | 28.6% (10/35) c | 80.0% (8/10) d | 14.3% (5/35) e |
| Total | 333 (90/90) a | 48.9% (44/90) b | 38.9% (35/90) c | 68.6% (24/35) d | 12.2% (11/90) e |
| Repair Mechanism | RNP (nM) | |||
|---|---|---|---|---|
| 73.5 | 147 | 220.6 | 294 | |
| NHEJ | 11.8% (10/85) a | 11.8% (10/85) a | 37.6% (32/85) a | 38.8% (33/85) a |
| MMEJ | 60.0% (3/5) b | 40.0% (2/5) b | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.; Yu, X.; Zhang, Z.; Tian, J.; Feng, N.; Tang, C.; Zou, G.; Zhang, J. An Efficient CRISPR/Cas9 Genome Editing System for a Ganoderma lucidum Cultivated Strain by Ribonucleoprotein Method. J. Fungi 2023, 9, 1170. https://doi.org/10.3390/jof9121170
Tan Y, Yu X, Zhang Z, Tian J, Feng N, Tang C, Zou G, Zhang J. An Efficient CRISPR/Cas9 Genome Editing System for a Ganoderma lucidum Cultivated Strain by Ribonucleoprotein Method. Journal of Fungi. 2023; 9(12):1170. https://doi.org/10.3390/jof9121170
Chicago/Turabian StyleTan, Yi, Xianglin Yu, Zhigang Zhang, Jialin Tian, Na Feng, Chuanhong Tang, Gen Zou, and Jingsong Zhang. 2023. "An Efficient CRISPR/Cas9 Genome Editing System for a Ganoderma lucidum Cultivated Strain by Ribonucleoprotein Method" Journal of Fungi 9, no. 12: 1170. https://doi.org/10.3390/jof9121170