
Functional Clustering of Metabolically Related Genes Is Conserved across Dikarya
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background and Introduction
2. Within Dikarya, Study of Ascomycetes Has Yielded Complex Insight into the Roles of Spatial Positioning on Gene Expression and Genome Organization
3. Basidiomycetes Are the Understudied Member of the Dikarya Clade
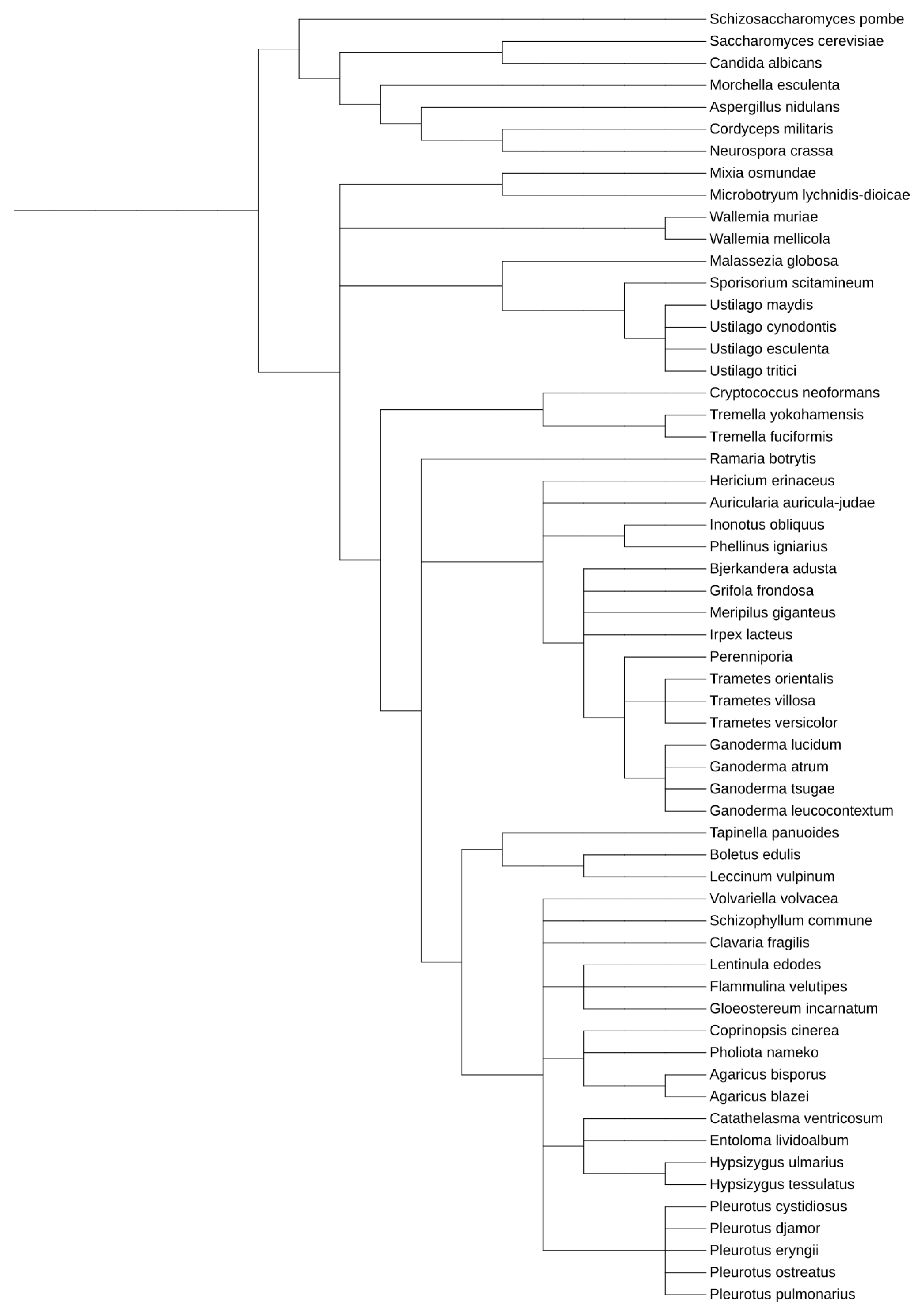
4. The Incidence and Prevalence of Functionally Related Gene Clusters across the Basidiomycetes Lineages
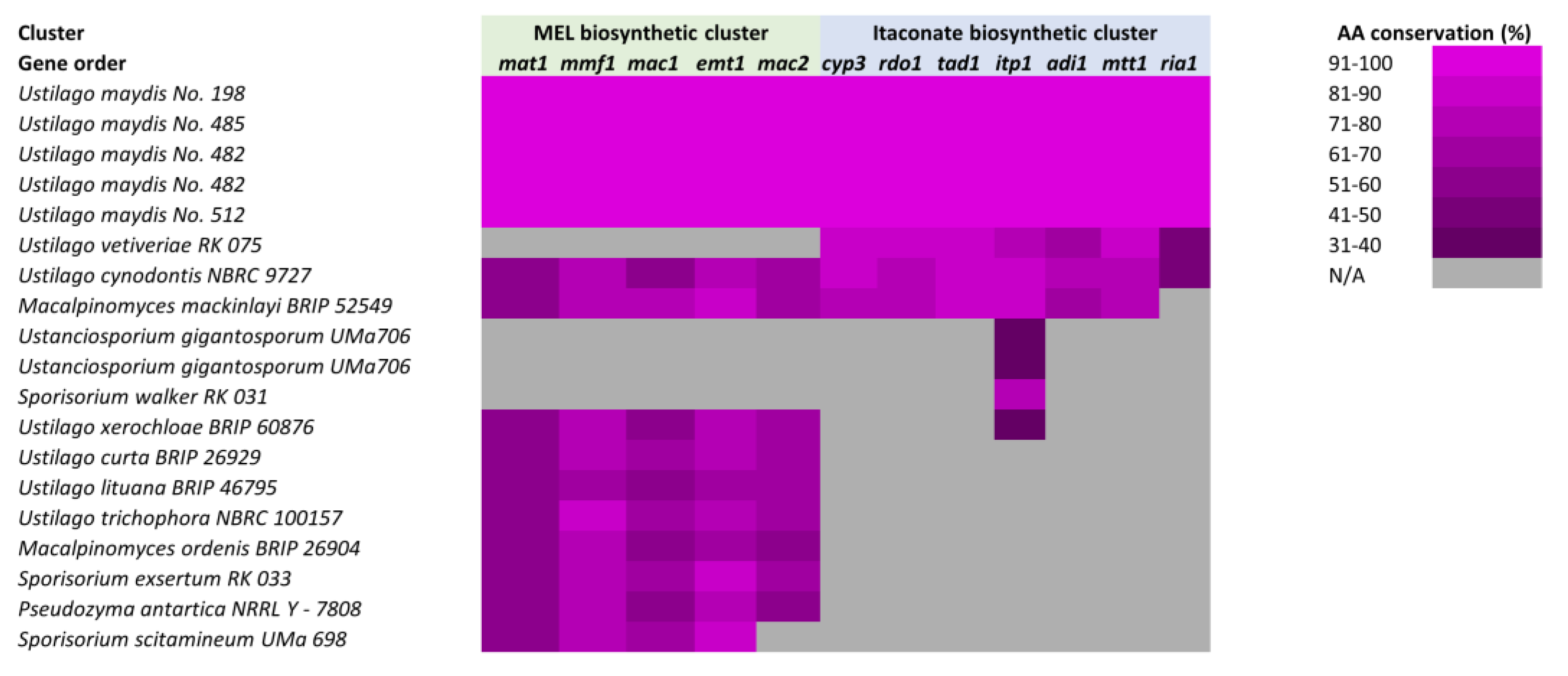
4.1. Ustilaginomycotina
4.2. Agaricomycotina
4.3. Pucciniomycotina
4.4. Wallemiomycotina
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stajich, J.E.; Berbee, M.L.; Blackwell, M.; Hibbett, D.S.; James, T.Y.; Spatafora, J.W.; Taylor, J.W. The Fungi. Curr. Biol. 2009, 19, R840–R845. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.; Weber, R. Introduction to Fungi, 3rd ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2007; ISBN 978-0-521-80739-5. [Google Scholar]
- Taylor, J.; Jacobson, D.; Fisher, M. The Evolution of Asexual Fungi: Reproduction, Speciation and Classification. Annu. Rev. Phytopathol. 1999, 37, 197–246. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Sun, S.; James, T.Y. Evolution of fungal sexual reproduction. Mycologia 2013, 105, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Heilmann-Clausen, J.; Barron, E.S.; Boddy, L.; Dahlberg, A.; Griffith, G.W.; Nordén, J.; Ovaskainen, O.; Perini, C.; Senn-Irlet, B.; Halme, P. A fungal perspective on conservation biology: Fungi and Conservation Biology. Conserv. Biol. 2015, 29, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sudbery, P. Candida albicans, a major human fungal pathogen. J. Microbiol. 2011, 49, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L. The fungal dimension of biodiversity: Magnitude, significance, and conservation. Mycol. Res. 1991, 95, 641–655. [Google Scholar] [CrossRef]
- Blackwell, M. The Fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef]
- Bisby, G.R.; Ainsworth, G.C. The numbers of fungi. Trans. Br. Mycol. Soc. 1943, 26, 16–19. [Google Scholar] [CrossRef]
- Martin, G. The Numbers of Fungi. Proc. Iowa Acad. Sci. 1951, 58, 175–178. [Google Scholar]
- Schmit, J.P.; Mueller, G.M. An estimate of the lower limit of global fungal diversity. Biodivers. Conserv. 2007, 16, 99–111. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 2017, 5, 5-4. [Google Scholar] [CrossRef] [PubMed]
- Locey, K.J.; Lennon, J.T. Scaling laws predict global microbial diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 5970–5975. [Google Scholar] [CrossRef] [PubMed]
- Boekhout, T.; Amend, A.S.; El Baidouri, F.; Gabaldón, T.; Geml, J.; Mittelbach, M.; Robert, V.; Tan, C.S.; Turchetti, B.; Vu, D.; et al. Trends in yeast diversity discovery. Fungal Divers. 2022, 114, 491–537. [Google Scholar] [CrossRef]
- Money, N.P. Fungal Diversity. In The Fungi; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–36. ISBN 978-0-12-382034-1. [Google Scholar]
- Picard, K.T. Coastal marine habitats harbor novel early-diverging fungal diversity. Fungal Ecol. 2017, 25, 1–13. [Google Scholar] [CrossRef]
- Manohar, C.S.; Raghukumar, C. Fungal diversity from various marine habitats deduced through culture-independent studies. FEMS Microbiol. Lett. 2013, 341, 69–78. [Google Scholar] [CrossRef]
- Lentendu, G.; Zinger, L.; Manel, S.; Coissac, E.; Choler, P.; Geremia, R.A.; Melodelima, C. Assessment of soil fungal diversity in different alpine tundra habitats by means of pyrosequencing. Fungal Divers. 2011, 49, 113–123. [Google Scholar] [CrossRef]
- Bass, D.; Howe, A.; Brown, N.; Barton, H.; Demidova, M.; Michelle, H.; Li, L.; Sanders, H.; Watkinson, S.C.; Willcock, S.; et al. Yeast forms dominate fungal diversity in the deep oceans. Proc. R. Soc. B. 2007, 274, 3069–3077. [Google Scholar] [CrossRef]
- Arenz, B.E.; Blanchette, R.A.; Farrell, R.L. Fungal Diversity in Antarctic Soils. In Antarctic Terrestrial Microbiology; Cowan, D.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 35–53. ISBN 978-3-642-45212-3. [Google Scholar]
- Chambergo, F.S.; Valencia, E.Y. Fungal biodiversity to biotechnology. Appl. Microbiol. Biotechnol. 2016, 100, 2567–2577. [Google Scholar] [CrossRef]
- McKelvey, S.M.; Murphy, R.A. Biotechnological Use of Fungal Enzymes. In Fungi; Kavanagh, K., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2017; pp. 201–225. ISBN 978-1-119-37431-2. [Google Scholar]
- Hyde, K.D.; Xu, J.; Rapior, S.; Jeewon, R.; Lumyong, S.; Niego, A.G.T.; Abeywickrama, P.D.; Aluthmuhandiram, J.V.S.; Brahamanage, R.S.; Brooks, S.; et al. The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers. 2019, 97, 1–136. [Google Scholar] [CrossRef]
- Almpani-Lekka, D.; Pfeiffer, S.; Schmidts, C.; Seo, S. A review on architecture with fungal biomaterials: The desired and the feasible. Fungal Biol. Biotechnol. 2021, 8, 17. [Google Scholar] [CrossRef]
- Dijksterhuis, J. Fungal spores: Highly variable and stress-resistant vehicles for distribution and spoilage. Food Microbiol. 2019, 81, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lücking, R.; et al. A higher-level phylogenetic classification of the Fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Ebersberger, I.; de Matos Simoes, R.; Kupczok, A.; Gube, M.; Kothe, E.; Voigt, K.; von Haeseler, A. A Consistent Phylogenetic Backbone for the Fungi. Mol. Biol. Evol. 2012, 29, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Xu, J.-W.; Xiao, J.-H.; Tang, Y.-J.; Xiao, H.; Zhong, J.-J. Cell Factories of Higher Fungi for Useful Metabolite Production. In Bioreactor Engineering Research and Industrial Applications I; Ye, Q., Bao, J., Zhong, J.-J., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 155, pp. 199–235. ISBN 978-3-662-49159-1. [Google Scholar]
- Taylor, J.W.; Bowman, B.H.; Berbee, M.L.; White, T.J. Fungal Model Organisms: Phylogenetics of Saccharomyces, Aspergillus, and Neurospora. Syst. Biol. 1993, 42, 440–457. [Google Scholar] [CrossRef]
- Kabir, M.A.; Hussain, M.A.; Ahmad, Z. Candida albicans: A Model Organism for Studying Fungal Pathogens. ISRN Microbiol. 2012, 2012, 538694. [Google Scholar] [CrossRef]
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An Ancient Yeast for Young Geneticists: A Primer on the Schizosaccharomyces pombe Model System. Genetics 2015, 201, 403–423. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef]
- Shen, X.-X.; Steenwyk, J.L.; LaBella, A.L.; Opulente, D.A.; Zhou, X.; Kominek, J.; Li, Y.; Groenewald, M.; Hittinger, C.T.; Rokas, A. Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci. Adv. 2020, 6, eabd0079. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hagee, D.; Abu Hardan, A.; Botero, J.; Arnone, J.T. Genomic clustering within functionally related gene families in Ascomycota fungi. Comput. Struct. Biotechnol. J. 2020, 18, 3267–3277. [Google Scholar] [CrossRef]
- Lofgren, L.A.; Uehling, J.K.; Branco, S.; Bruns, T.D.; Martin, F.; Kennedy, P.G. Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol. Ecol. 2019, 28, 721–730. [Google Scholar] [CrossRef]
- Vilgalys, R.; Gonzalez, D. Organization of ribosomal DNA in the basidiomycete Thanatephorus praticola. Curr. Genet. 1990, 18, 277–280. [Google Scholar] [CrossRef]
- Fraser, J.A.; Heitman, J. Fungal mating-type loci. Curr. Biol. 2003, 13, R792–R795. [Google Scholar] [CrossRef]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding Yeast for Budding Geneticists: A Primer on the Saccharomyces cerevisiae Model System. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef]
- Legrand, M.; Jaitly, P.; Feri, A.; d’Enfert, C.; Sanyal, K. Candida albicans: An Emerging Yeast Model to Study Eukaryotic Genome Plasticity. Trends Genet. 2019, 35, 292–307. [Google Scholar] [CrossRef]
- Piškur, J.; Langkjaer, R.B. Yeast genome sequencing: The power of comparative genomics: Yeast comparative genomics. Mol. Microbiol. 2004, 53, 381–389. [Google Scholar] [CrossRef]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic Expression Programs in the Response of Yeast Cells to Environmental Changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Tirosh, I.; Weinberger, A.; Carmi, M.; Barkai, N. A genetic signature of interspecies variations in gene expression. Nat. Genet. 2006, 38, 830–834. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef]
- Cohen, B.A.; Mitra, R.D.; Hughes, J.D.; Church, G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000, 26, 183–186. [Google Scholar] [CrossRef]
- Douglas, H.C.; Hawthorne, D.C. Regulation of genes controlling synthesis of the galactose pathway enzymes in yeast. Genetics 1966, 54, 911–916. [Google Scholar] [CrossRef]
- John, T.P.S.; Davis, R.W. The organization and transcription of the galactose gene cluster of Saccharomyces. J. Mol. Biol. 1981, 152, 285–315. [Google Scholar] [CrossRef]
- John, T.P.S.; Scherer, S.; McDonell, M.W.; Davis, R.W. Deletion analysis of the Saccharomyces GAL gene cluster. J. Mol. Biol. 1981, 152, 317–334. [Google Scholar] [CrossRef]
- Johnston, M. A model fungal gene regulatory mechanism: The GAL genes of Saccharomyces cerevisiae. Microbiol. Rev. 1987, 51, 458–476. [Google Scholar] [CrossRef]
- Kundu, S.; Horn, P.J.; Peterson, C.L. SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev. 2007, 21, 997–1004. [Google Scholar] [CrossRef]
- Douglas, H.C.; Hawthorne, D.C. Enzymatic expression and genetic linkage of genes controlling galactose utilization in saccharomyces. Genetics 1964, 49, 837–844. [Google Scholar] [CrossRef]
- McGary, K.L.; Slot, J.C.; Rokas, A. Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc. Natl. Acad. Sci. USA 2013, 110, 11481–11486. [Google Scholar] [CrossRef]
- Wade, C.H.; Umbarger, M.A.; McAlear, M.A. The budding yeast rRNA and ribosome biosynthesis (RRB) regulon contains over 200 genes. Yeast 2006, 23, 293–306. [Google Scholar] [CrossRef]
- Arnone, J.T.; McAlear, M.A. Adjacent Gene Pairing Plays a Role in the Coordinated Expression of Ribosome Biogenesis Genes MPP10 and YJR003C in Saccharomyces cerevisiae. Eukaryot. Cell 2011, 10, 43–53. [Google Scholar] [CrossRef]
- Arnone, J.T.; Robbins-Pianka, A.; Arace, J.R.; Kass-Gergi, S.; McAlear, M.A. The adjacent positioning of co-regulated gene pairs is widely conserved across eukaryotes. BMC Genom. 2012, 13, 546. [Google Scholar] [CrossRef]
- Arnone, J.T.; Arace, J.R.; Soorneedi, A.R.; Citino, T.T.; Kamitaki, T.L.; McAlear, M.A. Dissecting the cis and trans Elements That Regulate Adjacent-Gene Coregulation in Saccharomyces cerevisiae. Eukaryot. Cell 2014, 13, 738–748. [Google Scholar] [CrossRef]
- Warner, J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999, 24, 437–440. [Google Scholar] [CrossRef]
- Osbourn, A.E.; Field, B. Operons. Cell. Mol. Life Sci. 2009, 66, 3755–3775. [Google Scholar] [CrossRef]
- Eldabagh, R.S.; Mejia, N.G.; Barrett, R.L.; Monzo, C.R.; So, M.K.; Foley, J.J.; Arnone, J.T. Systematic Identification, Characterization, and Conservation of Adjacent-Gene Coregulation in the Budding Yeast Saccharomyces cerevisiae. mSphere 2018, 3, e00220-18. [Google Scholar] [CrossRef]
- Asfare, S.; Eldabagh, R.; Siddiqui, K.; Patel, B.; Kaba, D.; Mullane, J.; Siddiqui, U.; Arnone, J.T. Systematic Analysis of Functionally Related Gene Clusters in the Opportunistic Pathogen, Candida albicans. Microorganisms 2021, 9, 276. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef]
- Kellis, M.; Birren, B.W.; Lander, E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 2004, 428, 617–624. [Google Scholar] [CrossRef]
- Chu, A.M.; Davis, R.W. High-Throughput Creation of a Whole-Genome Collection of Yeast Knockout Strains. In Microbial Gene Essentiality: Protocols and Bioinformatics; Osterman, A.L., Gerdes, S.Y., Eds.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, USA, 2008; Volume 416, pp. 205–220. ISBN 978-1-58829-378-7. [Google Scholar]
- Ben-Shitrit, T.; Yosef, N.; Shemesh, K.; Sharan, R.; Ruppin, E.; Kupiec, M. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat. Methods 2012, 9, 373–378. [Google Scholar] [CrossRef]
- Atias, N.; Kupiec, M.; Sharan, R. Systematic identification and correction of annotation errors in the genetic interaction map of Saccharomyces cerevisiae. Nucleic Acids Res. 2016, 44, e50. [Google Scholar] [CrossRef]
- Elgin, S.C.R.; Reuter, G. Position-Effect Variegation, Heterochromatin Formation, and Gene Silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef]
- Renauld, H.; Aparicio, O.M.; Zierath, P.D.; Billington, B.L.; Chhablani, S.K.; Gottschling, D.E. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev. 1993, 7, 1133–1145. [Google Scholar] [CrossRef]
- Ottaviani, A.; Gilson, E.; Magdinier, F. Telomeric position effect: From the yeast paradigm to human pathologies? Biochimie 2008, 90, 93–107. [Google Scholar] [CrossRef]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Huh, W.-K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J. The Genomic Landscape of Position Effects on Protein Expression Level and Noise in Yeast. Cell Syst. 2016, 2, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-L.; Li, B.-Z.; Zhang, W.-Z.; Song, K.; Qi, H.; Dai, J.; Yuan, Y.-J. Genome-wide landscape of position effects on heterogeneous gene expression in Saccharomyces cerevisiae. Biotechnol. Biofuels 2017, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Cadena, P.; Sternberg, P.W. Enhancer Sharing Promotes Neighborhoods of Transcriptional Regulation across Eukaryotes. G3 Genes Genomes Genet. 2016, 6, 4167–4174. [Google Scholar] [CrossRef]
- Cera, A.; Holganza, M.K.; Hardan, A.A.; Gamarra, I.; Eldabagh, R.S.; Deschaine, M.; Elkamhawy, S.; Sisso, E.M.; Foley, J.J.; Arnone, J.T. Functionally Related Genes Cluster into Genomic Regions That Coordinate Transcription at a Distance in Saccharomyces cerevisiae. mSphere 2019, 4, e00063-19. [Google Scholar] [CrossRef] [PubMed]
- Arnone, J.T. Genomic Considerations for the Modification of Saccharomyces cerevisiae for Biofuel and Metabolite Biosynthesis. Microorganisms 2020, 8, 321. [Google Scholar] [CrossRef]
- Bentil, J.A. Biocatalytic potential of basidiomycetes: Relevance, challenges and research interventions in industrial processes. Sci. Afr. 2021, 11, e00717. [Google Scholar] [CrossRef]
- Wijayawardene, N.N.; Hyde, K.D.; Al-Ani, L.K.T.; Tedersoo, L.; Haelewaters, D.; Rajeshkumar, K.C.; Zhao, R.L.; Aptroot, A.; Leontyev, D.V.; Saxena, R.K.; et al. Outline of Fungi and fungus-like taxa. Mycosphere 2020, 11, 1060–1456. [Google Scholar] [CrossRef]
- Schwarze, F.W.M.R. Wood decay under the microscope. Fungal Biol. Rev. 2007, 21, 133–170. [Google Scholar] [CrossRef]
- He, M.-Q.; Zhao, R.-L.; Liu, D.-M.; Denchev, T.T.; Begerow, D.; Yurkov, A.; Kemler, M.; Millanes, A.M.; Wedin, M.; McTaggart, A.R.; et al. Species diversity of Basidiomycota. Fungal Divers. 2022, 114, 281–325. [Google Scholar] [CrossRef]
- Morrow, C.A.; Fraser, J.A. Sexual reproduction and dimorphism in the pathogenic basidiomycetes. FEMS Yeast Res. 2009, 9, 161–177. [Google Scholar] [CrossRef]
- Tsujiyama, S.; Ueno, H. Performance of wood-rotting fungi-based enzymes on enzymic saccharification of rice straw: Saccharification of rice straw by fungal enzymes. J. Sci. Food Agric. 2013, 93, 2841–2848. [Google Scholar] [CrossRef]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-Polysaccharide-Degrading Enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef]
- Uber, T.M.; Backes, E.; Saute, V.M.S.; da Silva, B.P.; Corrêa, R.C.G.; Kato, C.G.; Seixas, F.A.V.; Bracht, A.; Peralta, R.M. Enzymes from basidiomycetes—Peculiar and efficient tools for biotechnology. In Biotechnology of Microbial Enzymes; Elsevier: Amsterdam, The Netherlands, 2023; pp. 129–164. ISBN 978-0-443-19059-9. [Google Scholar]
- Sánchez, C. Lignocellulosic residues: Biodegradation and bioconversion by fungi. Biotechnol. Adv. 2009, 27, 185–194. [Google Scholar] [CrossRef]
- Tian, M.; Zhao, P.; Li, G.; Zhang, K. In Depth Natural Product Discovery from the Basidiomycetes Stereum Species. Microorganisms 2020, 8, 1049. [Google Scholar] [CrossRef]
- Shankar, A.; Sharma, K.K. Fungal secondary metabolites in food and pharmaceuticals in the era of multi-omics. Appl. Microbiol. Biotechnol. 2022, 106, 3465–3488. [Google Scholar] [CrossRef]
- Zhao, R.-L.; Li, G.-J.; Sánchez-Ramírez, S.; Stata, M.; Yang, Z.-L.; Wu, G.; Dai, Y.-C.; He, S.-H.; Cui, B.-K.; Zhou, J.-L.; et al. A six-gene phylogenetic overview of Basidiomycota and allied phyla with estimated divergence times of higher taxa and a phyloproteomics perspective. Fungal Divers. 2017, 84, 43–74. [Google Scholar] [CrossRef]
- Sandargo, B.; Chepkirui, C.; Cheng, T.; Chaverra-Muñoz, L.; Thongbai, B.; Stadler, M.; Hüttel, S. Biological and chemical diversity go hand in hand: Basidiomycota as source of new pharmaceuticals and agrochemicals. Biotechnol. Adv. 2019, 37, 107344. [Google Scholar] [CrossRef] [PubMed]
- Begerow, D.; Stoll, M.; Bauer, R. A phylogenetic hypothesis of Ustilaginomycotina based on multiple gene analyses and morphological data. Mycologia 2006, 98, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Brefort, T.; Doehlemann, G.; Mendoza-Mendoza, A.; Reissmann, S.; Djamei, A.; Kahmann, R. Ustilago maydis as a Pathogen. Annu. Rev. Phytopathol. 2009, 47, 423–445. [Google Scholar] [CrossRef] [PubMed]
- Banuett, F. Genetics of Ustilago maydis, a fungal pathogen that induces tumors in maize. Annu. Rev. Genet. 1995, 29, 179–208. [Google Scholar] [CrossRef]
- Doebley, J. The Genetics of Maize Evolution. Annu. Rev. Genet. 2004, 38, 37–59. [Google Scholar] [CrossRef]
- Mueller, O.; Kahmann, R.; Aguilar, G.; Trejo-Aguilar, B.; Wu, A.; de Vries, R.P. The secretome of the maize pathogen Ustilago maydis. Fungal Genet. Biol. 2008, 45, S63–S70. [Google Scholar] [CrossRef]
- Mendgen, K.; Hahn, M. Plant infection and the establishment of fungal biotrophy. Trends Plant Sci. 2002, 7, 352–356. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.-J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Brefort, T.; Tanaka, S.; Neidig, N.; Doehlemann, G.; Vincon, V.; Kahmann, R. Characterization of the largest effector gene cluster of Ustilago maydis. PLoS Pathog. 2014, 10, e1003866. [Google Scholar] [CrossRef]
- Basse, C.W.; Kolb, S.; Kahmann, R. A maize-specifically expressed gene cluster in Ustilago maydis. Mol. Microbiol. 2002, 43, 75–93. [Google Scholar] [CrossRef]
- Coelho, A.L.S.; Feuser, P.E.; Carciofi, B.A.M.; de Andrade, C.J.; de Oliveira, D. Mannosylerythritol lipids: Antimicrobial and biomedical properties. Appl. Microbiol. Biotechnol. 2020, 104, 2297–2318. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, Z.; Zeng, G.; Zhong, H.; Liu, Y.; Jiang, Y.; Li, M.; He, X.; He, Y. Characteristics of mannosylerythritol lipids and their environmental potential. Carbohydr. Res. 2015, 407, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hewald, S.; Linne, U.; Scherer, M.; Marahiel, M.A.; Kämper, J.; Bölker, M. Identification of a gene cluster for biosynthesis of mannosylerythritol lipids in the basidiomycetous fungus Ustilago maydis. Appl. Environ. Microbiol. 2006, 72, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Haas, H. Molecular genetics of fungal siderophore biosynthesis and uptake: The role of siderophores in iron uptake and storage. Appl. Microbiol. Biotechnol. 2003, 62, 316–330. [Google Scholar] [CrossRef]
- Mei, B.; Budde, A.D.; Leong, S.A. sid1, a gene initiating siderophore biosynthesis in Ustilago maydis: Molecular characterization, regulation by iron, and role in phytopathogenicity. Proc. Natl. Acad. Sci. USA 1993, 90, 903–907. [Google Scholar] [CrossRef]
- Yuan, W.M.; Gentil, G.D.; Budde, A.D.; Leong, S.A. Characterization of the Ustilago maydis sid2 gene, encoding a multidomain peptide synthetase in the ferrichrome biosynthetic gene cluster. J. Bacteriol. 2001, 183, 4040–4051. [Google Scholar] [CrossRef]
- Haskins, R.H.; Thorn, J.A. Biochemistry of the ustilaginales: VII. Antibiotic activity of ustilagic acid. Can. J. Bot. 1951, 29, 585–592. [Google Scholar] [CrossRef]
- Teichmann, B.; Linne, U.; Hewald, S.; Marahiel, M.A.; Bölker, M. A biosynthetic gene cluster for a secreted cellobiose lipid with antifungal activity from Ustilago maydis. Mol. Microbiol. 2007, 66, 525–533. [Google Scholar] [CrossRef]
- Teichmann, B.; Liu, L.; Schink, K.O.; Bölker, M. Activation of the ustilagic acid biosynthesis gene cluster in Ustilago maydis by the C2H2 zinc finger transcription factor Rua1. Appl. Environ. Microbiol. 2010, 76, 2633–2640. [Google Scholar] [CrossRef]
- Willke, T.; Vorlop, K.-D. Biotechnological production of itaconic acid. Appl. Microbiol. Biotechnol. 2001, 56, 289–295. [Google Scholar] [CrossRef]
- Geiser, E.; Przybilla, S.K.; Friedrich, A.; Buckel, W.; Wierckx, N.; Blank, L.M.; Bölker, M. Ustilago maydis produces itaconic acid via the unusual intermediate trans-aconitate. Microb. Biotechnol. 2016, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Zambanini, T.; Hartmann, S.K.; Schmitz, L.M.; Büttner, L.; Tehrani, H.H.; Geiser, E.; Beudels, M.; Venc, D.; Wandrey, G.; Büchs, J.; et al. Promoters from the itaconate cluster of Ustilago maydis are induced by nitrogen depletion. Fungal Biol. Biotechnol. 2017, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Grendelmeier, P.; Scheynius, A.; Crameri, R. The Role of Sensitization to Malassezia sympodialis in Atopic Eczema. In Chemical Immunology and Allergy; Crameri, R., Ed.; KARGER: Basel, Switzerland, 2006; pp. 98–109. ISBN 978-3-8055-8000-7. [Google Scholar]
- Xu, J.; Saunders, C.W.; Hu, P.; Grant, R.A.; Boekhout, T.; Kuramae, E.E.; Kronstad, J.W.; DeAngelis, Y.M.; Reeder, N.L.; Johnstone, K.R.; et al. Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc. Natl. Acad. Sci. USA 2007, 104, 18730–18735. [Google Scholar] [CrossRef] [PubMed]
- Oraby, A.; Werner, N.; Sungur, Z.; Zibek, S. Factors Affecting the Synthesis of Cellobiose Lipids by Sporisorium scitamineum. Front. Bioeng. Biotechnol. 2020, 8, 555647. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.S. A phylogenetic overview of the Agaricomycotina. Mycologia 2006, 98, 917–925. [Google Scholar] [CrossRef]
- Sánchez-García, M.; Ryberg, M.; Khan, F.K.; Varga, T.; Nagy, L.G.; Hibbett, D.S. Fruiting body form, not nutritional mode, is the major driver of diversification in mushroom-forming fungi. Proc. Natl. Acad. Sci. USA 2020, 117, 32528–32534. [Google Scholar] [CrossRef]
- Slot, J.C.; Rokas, A. Multiple GAL pathway gene clusters evolved independently and by different mechanisms in fungi. Proc. Natl. Acad. Sci. USA 2010, 107, 10136–10141. [Google Scholar] [CrossRef]
- Harrison, M.-C.; LaBella, A.L.; Hittinger, C.T.; Rokas, A. The evolution of the GALactose utilization pathway in budding yeasts. Trends Genet. 2022, 38, 97–106. [Google Scholar] [CrossRef]
- Ye, L.; Wang, S.; Zheng, J.; Chen, L.; Shen, L.; Kuang, Y.; Wang, Y.; Peng, Y.; Hu, C.; Wang, L.; et al. Functional Characterization of the GlcNAc Catabolic Pathway in Cryptococcus deneoformans. Appl. Environ. Microbiol. 2022, 88, e00437-22. [Google Scholar] [CrossRef]
- Lian, T.; Simmer, M.I.; D’Souza, C.A.; Steen, B.R.; Zuyderduyn, S.D.; Jones, S.J.M.; Marra, M.A.; Kronstad, J.W. Iron-regulated transcription and capsule formation in the fungal pathogen Cryptococcus neoformans: Iron regulation in C. neoformans. Mol. Microbiol. 2004, 55, 1452–1472. [Google Scholar] [CrossRef]
- Doddapaneni, H.; Chakraborty, R.; Yadav, J.S. Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: Evidence for gene duplications and extensive gene clustering. BMC Genom. 2005, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Covert, S.F.; Wymelenberg, A.V.; Cullen, D. Structure, organization, and transcription of a cellobiohydrolase gene cluster from Phanerochaete chrysosporium. Appl. Environ. Microbiol. 1992, 58, 2168–2175. [Google Scholar] [CrossRef] [PubMed]
- Larrondo, L.F.; González, B.; Cullen, D.; Vicuña, R. Characterization of a multicopper oxidase gene cluster in Phanerochaete chrysosporium and evidence of altered splicing of the mco transcripts. Microbiology 2004, 150, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.; Cullen, D. Organization and Differential Regulation of a Cluster of Lignin Peroxidase Genes of Phanerochaete chrysosporium. J. Bacteriol. 1999, 181, 3427–3432. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.S.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Bouhired, S.; Hoffmeister, D. Characterization of the atromentin biosynthesis genes and enzymes in the homobasidiomycete Tapinella panuoides. Fungal Genet. Biol. 2008, 45, 1487–1496. [Google Scholar] [CrossRef]
- Khanna, J.M.; Malone, M.H.; Euler, K.L.; Brady, L.R. Atromentin. Anticoagulant from Hydnellum diabolus. J. Pharm. Sci. 1965, 54, 1016–1020. [Google Scholar] [CrossRef]
- Sullivan, G.; Guess, W.L. Atromentin: A smooth muscle stimulant in Clitocybe subilludens. Lloydia 1969, 32, 72–75. [Google Scholar]
- Boh, B.; Berovic, M.; Zhang, J.; Lin, Z.-B. Ganoderma lucidum and its pharmaceutically active compounds. In Biotechnology Annual Review; Elsevier: Amsterdam, The Netherlands, 2007; Volume 13, pp. 265–301. ISBN 978-0-444-53032-5. [Google Scholar]
- Sanodiya, B.; Thakur, G.; Baghel, R.; Prasad, G.; Bisen, P. Ganoderma lucidum: A Potent Pharmacological Macrofungus. Curr. Pharm. Biotechnol. 2009, 10, 717–742. [Google Scholar] [CrossRef]
- Sliva, D. Ganoderma lucidum (Reishi) in cancer treatment. Integr Cancer Ther. 2003, 2, 358–364. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed]
- Mapoung, S.; Umsumarng, S.; Semmarath, W.; Arjsri, P.; Thippraphan, P.; Yodkeeree, S.; Limtrakul, P. Skin Wound-Healing Potential of Polysaccharides from Medicinal Mushroom Auricularia auricula-judae (Bull.). J. Fungi 2021, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Fukushima-Sakuno, E. Bioactive small secondary metabolites from the mushrooms Lentinula edodes and Flammulina velutipes. J. Antibiot. 2020, 73, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Park, Y.-J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.-S.; Park, H.-R.; Yoon, D.-E.; Nam, J.-Y.; et al. Whole Genome and Global Gene Expression Analyses of the Model Mushroom Flammulina velutipes Reveal a High Capacity for Lignocellulose Degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Ferreira, D.S.S.; Kato, R.B.; Miranda, F.M.; da Costa Pinheiro, K.; Fonseca, P.L.C.; Tomé, L.M.R.; Vaz, A.B.M.; Badotti, F.; Ramos, R.T.J.; Brenig, B.; et al. Draft genome sequence of Trametes villosa (Sw.) Kreisel CCMB561, a tropical white-rot Basidiomycota from the semiarid region of Brazil. Data Brief 2018, 18, 1581–1587. [Google Scholar] [CrossRef]
- Chiku, K.; Sugiyama, S.; Akimoto, K.; Yoshida, M. Draft Genome Sequences of Two Basidiomycetous Yeasts, Tremella yokohamensis and Tremella fuciformis. Microbiol. Resour. Announc. 2022, 11, e00573-22. [Google Scholar] [CrossRef]
- Aime, M.C.; Matheny, P.B.; Henk, D.A.; Frieders, E.M.; Nilsson, R.H.; Piepenbring, M.; McLaughlin, D.J.; Szabo, L.J.; Begerow, D.; Sampaio, J.P.; et al. An overview of the higher level classification of Pucciniomycotina based on combined analyses of nuclear large and small subunit rDNA sequences. Mycologia 2006, 98, 896–905. [Google Scholar] [CrossRef]
- Schell, W.A.; Lee, A.G.; Aime, M.C. A new lineage in Pucciniomycotina: Class Tritirachiomycetes, order Tritirachiales, family Tritirachiaceae. Mycologia 2011, 103, 1331–1340. [Google Scholar] [CrossRef]
- Toome, M.; Roberson, R.W.; Aime, M.C. Meredithblackwellia eburnea gen. et sp. nov., Kriegeriaceae fam. nov. and Kriegeriales ord. nov—Toward resolving higher-level classification in Microbotryomycetes. Mycologia 2013, 105, 486–495. [Google Scholar] [CrossRef]
- Toome, M.; Ohm, R.A.; Riley, R.W.; James, T.Y.; Lazarus, K.L.; Henrissat, B.; Albu, S.; Boyd, A.; Chow, J.; Clum, A.; et al. Genome sequencing provides insight into the reproductive biology, nutritional mode and ploidy of the fern pathogen Mixia osmundae. New Phytol. 2014, 202, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, S.; Cuomo, C.A.; Lin, Y.-C.; Aerts, A.; Tisserant, E.; Veneault-Fourrey, C.; Joly, D.L.; Hacquard, S.; Amselem, J.; Cantarel, B.L.; et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA 2011, 108, 9166–9171. [Google Scholar] [CrossRef] [PubMed]
- Alexander, H.M. An experimental field study of anther-smut disease of silene alba caused by Ustilago violacea: Genotypic variation and disease incidence. Evolution 1989, 43, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.F.; Perlin, M.H. Differential expression of mepA, mepC and smtE during growth and development of Microbotryum violaceum. Mycologia 2005, 97, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Perlin, M.H.; Amselem, J.; Fontanillas, E.; Toh, S.S.; Chen, Z.; Goldberg, J.; Duplessis, S.; Henrissat, B.; Young, S.; Zeng, Q.; et al. Sex and parasites: Genomic and transcriptomic analysis of Microbotryum lychnidis-dioicae, the biotrophic and plant-castrating anther smut fungus. BMC Genom. 2015, 16, 461. [Google Scholar] [CrossRef]
- Sugiyama, J.; Katumoto, K. Identity of the plasmodial slime mold Phytoceratiomyxa osmundae and the lectotypification of Taphrina osmundae, the basionym of Mixia osmundae. Mycoscience 2008, 49, 192–198. [Google Scholar] [CrossRef]
- Sugiyama, J.; Nishida, H.; Hosoya, T.; Kakishima, M. The enigmatic Mixia osmundae revisited: A systematic review including new distributional data and recent advances in its phylogeny and phylogenomics. Mycologia 2018, 110, 179–191. [Google Scholar] [CrossRef]
- Mix, A.J. Taphrina osmundae Nishida and Taphrina higginsii sp. nov. Mycologia 1947, 39, 71–76. [Google Scholar] [CrossRef]
- Zalar, P.; de Hoog, G.S.; Schroers, H.-J.; Frank, J.M.; Gunde-Cimerman, N. Taxonomy and phylogeny of the xerophilic genus Wallemia (Wallemiomycetes and Wallemiales, cl. et ord. nov.). Antonie Van Leeuwenhoek 2005, 87, 311–328. [Google Scholar] [CrossRef]
- Zajc, J.; Kogej, T.; Galinski, E.A.; Ramos, J.; Gunde-Cimerman, N. Osmoadaptation strategy of the most halophilic fungus, Wallemia ichthyophaga, growing optimally at salinities above 15% NaCl. Appl. Environ. Microbiol. 2014, 80, 247–256. [Google Scholar] [CrossRef]
- Gostinčar, C.; Zalar, P.; Gunde-Cimerman, N. No need for speed: Slow development of fungi in extreme environments. Fungal Biol. Rev. 2022, 39, 1–14. [Google Scholar] [CrossRef]
- Zajc, J.; Gunde-Cimerman, N. The Genus Wallemia—From Contamination of Food to Health Threat. Microorganisms 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Reboux, G.; Piarroux, R.; Mauny, F.; Madroszyk, A.; Millon, L.; Bardonnet, K.; Dalphin, J.-C. Role of Molds in Farmer’s Lung Disease in Eastern France. Am. J. Respir. Crit. Care Med. 2001, 163, 1534–1539. [Google Scholar] [CrossRef]
- Lacasse, Y.; Cormier, Y. Hypersensitivity pneumonitis. Orphanet J. Rare Dis. 2006, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gostinčar, C.; Fang, C.; Zajc, J.; Hou, Y.; Song, Z.; Gunde-Cimerman, N. Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola. Genes 2019, 10, 427. [Google Scholar] [CrossRef]
- Padamsee, M.; Kumar, T.K.A.; Riley, R.; Binder, M.; Boyd, A.; Calvo, A.M.; Furukawa, K.; Hesse, C.; Hohmann, S.; James, T.Y.; et al. The genome of the xerotolerant mold Wallemia sebi reveals adaptations to osmotic stress and suggests cryptic sexual reproduction. Fungal Genet. Biol. 2012, 49, 217–226. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Vilar, J.M.G.; Guet, C.C.; Leibler, S. Modeling network dynamics: The lac operon, a case study. J. Cell Biol. 2003, 161, 471–476. [Google Scholar] [CrossRef]
- Blumenthal, T.; Evans, D.; Link, C.D.; Guffanti, A.; Lawson, D.; Thierry-Mieg, J.; Thierry-Mieg, D.; Chiu, W.L.; Duke, K.; Kiraly, M.; et al. A global analysis of Caenorhabditis elegans operons. Nature 2002, 417, 851–854. [Google Scholar] [CrossRef]
- Blumenthal, T.; Gleason, K.S. Caenorhabditis elegans operons: Form and function. Nat. Rev. Genet. 2003, 4, 110–118. [Google Scholar] [CrossRef]
- Schmidt-Dannert, C. Biocatalytic portfolio of Basidiomycota. Curr. Opin. Chem. Biol. 2016, 31, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, L.; Wibberg, D.; Busche, T.; Rückert, C.; Müsgens, A.; Kalinowski, J.; Blank, L.M. Seventeen Ustilaginaceae High-Quality Genome Sequences Allow Phylogenomic Analysis and Provide Insights into Secondary Metabolite Synthesis. J. Fungi 2022, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.P.; Wolfe, K.H. The Yeast Gene Order Browser: Combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 2005, 15, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.P. Visualizing syntenic relationships among the hemiascomycetes with the Yeast Gene Order Browser. Nucleic Acids Res. 2006, 34, D452–D455. [Google Scholar] [CrossRef] [PubMed]
- Maguire, S.L.; ÓhÉigeartaigh, S.S.; Byrne, K.P.; Schröder, M.S.; O’Gaora, P.; Wolfe, K.H.; Butler, G. Comparative Genome Analysis and Gene Finding in Candida Species Using CGOB. Mol. Biol. Evol. 2013, 30, 1281–1291. [Google Scholar] [CrossRef]
- Douglass, A.P.; Byrne, K.P.; Wolfe, K.H. The Methylotroph Gene Order Browser (MGOB) reveals conserved synteny and ancestral centromere locations in the yeast family Pichiaceae. FEMS Yeast Res. 2019, 19, foz058. [Google Scholar] [CrossRef]
- McGowan, J.; Byrne, K.P.; Fitzpatrick, D.A. Comparative Analysis of Oomycete Genome Evolution Using the Oomycete Gene Order Browser (OGOB). Genome Biol. Evol. 2019, 11, 189–206. [Google Scholar] [CrossRef]
- Kjærbølling, I.; Vesth, T.; Andersen, M.R. Resistance Gene-Directed Genome Mining of 50 Aspergillus Species. Msystems 2019, 4, e00085-19. [Google Scholar] [CrossRef]
- Marcet-Houben, M.; Gabaldón, T. Evolutionary and functional patterns of shared gene neighbourhood in fungi. Nat. Microbiol. 2019, 4, 2383–2392. [Google Scholar] [CrossRef]
- Slot, J.C.; Gluck-Thaler, E. Metabolic gene clusters, fungal diversity, and the generation of accessory functions. Curr. Opin. Genet. Dev. 2019, 58–59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Wisecaver, J.H.; Slot, J.C.; Rokas, A. The Evolution of Fungal Metabolic Pathways. PLoS Genet. 2014, 10, e1004816. [Google Scholar] [CrossRef] [PubMed]
- Nützmann, H.-W.; Scazzocchio, C.; Osbourn, A. Metabolic Gene Clusters in Eukaryotes. Annu. Rev. Genet. 2018, 52, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Kathuria, S.; Agarwal, K.; Meis, J.F. Recognizing Filamentous Basidiomycetes as Agents of Human Disease: A Review. Med. Mycol. 2014, 52, 782–797. [Google Scholar] [CrossRef]
- Skalski, J.H.; Limon, J.J.; Sharma, P.; Gargus, M.D.; Nguyen, C.; Tang, J.; Coelho, A.L.; Hogaboam, C.M.; Crother, T.R.; Underhill, D.M. Expansion of Commensal Fungus Wallemia mellicola in the Gastrointestinal Mycobiota Enhances the Severity of Allergic Airway Disease in Mice. PLoS Pathog. 2018, 14, e1007260. [Google Scholar] [CrossRef]
- Gomes, D.C.V.; de Alencar, M.V.O.B.; Dos Reis, A.C.; de Lima, R.M.T.; de Oliveira Santos, J.V.; da Mata, A.M.O.F.; Dias, A.C.S.; da Costa Junior, J.S.; de Medeiros, M.D.G.F.; Paz, M.F.C.J.; et al. Antioxidant, Anti-Inflammatory and Cytotoxic/Antitumoral Bioactives from the Phylum Basidiomycota and Their Possible Mechanisms of Action. Biomed. Pharmacother. 2019, 112, 108643. [Google Scholar] [CrossRef]
- Van der Torre, M.H.; Andrews, R.A.; Hooker, E.; Rankin, A.; Dodd, S. Systematic review on Cryptococcus neoformans/Cryptococcus gattii species complex infections with recommendations for practice in health and care settings. Clin. Infect. Pract. 2022, 15, 100154. [Google Scholar] [CrossRef]
- Silva, C.; Juan-Sallés, C.; Mendes, J.; Mendes, A.; Ruivo, M.; Abad, J.L.; Hagen, F.; Colom, M.F. Cryptococcus bacillisporus Causing Cryptococcoma of the Beak of an African Grey Parrot (Psittacus erithacus), Portugal. Med. Mycol. Case Rep. 2021, 34, 8–12. [Google Scholar] [CrossRef]
- Wang, Z.H.; Yan, N.; Luo, X.; Guo, S.S.; Xue, S.Q.; Liu, J.Q.; Zhang, J.Z.; Guo, D.P. Gene Expression in the Smut Fungus Ustilago esculenta Governs Swollen Gall Metamorphosis in Zizania latifolia. Microb. Pathog. 2020, 143, 104107. [Google Scholar] [CrossRef]
- Quijano, C.D.; Wichmann, F.; Schlaich, T.; Fammartino, A.; Huckauf, J.; Schmidt, K.; Unger, C.; Broer, I.; Sautter, C. KP4 to Control Ustilago tritici in Wheat: Enhanced Greenhouse Resistance to Loose Smut and Changes in Transcript Abundance of Pathogen Related Genes in Infected KP4 Plants. Biotechnol. Rep. 2016, 11, 90–98. [Google Scholar] [CrossRef]
- Tehrani, H.H.; Tharmasothirajan, A.; Track, E.; Blank, L.M.; Wierckx, N. Engineering the Morphology and Metabolism of PH Tolerant Ustilago cynodontis for Efficient Itaconic Acid Production. Metab. Eng. 2019, 54, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kunčič, M.K.; Zajc, J.; Drobne, D.; Tkalec, Ž.P.; Gunde-Cimerman, N. Morphological Responses to High Sugar Concentrations Differ from Adaptation to High Salt Concentrations in the Xerophilic Fungi Wallemia spp. Fungal Biol. 2013, 117, 466–478. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cittadino, G.M.; Andrews, J.; Purewal, H.; Estanislao Acuña Avila, P.; Arnone, J.T. Functional Clustering of Metabolically Related Genes Is Conserved across Dikarya. J. Fungi 2023, 9, 523. https://doi.org/10.3390/jof9050523
Cittadino GM, Andrews J, Purewal H, Estanislao Acuña Avila P, Arnone JT. Functional Clustering of Metabolically Related Genes Is Conserved across Dikarya. Journal of Fungi. 2023; 9(5):523. https://doi.org/10.3390/jof9050523
Chicago/Turabian StyleCittadino, Gina M., Johnathan Andrews, Harpreet Purewal, Pedro Estanislao Acuña Avila, and James T. Arnone. 2023. "Functional Clustering of Metabolically Related Genes Is Conserved across Dikarya" Journal of Fungi 9, no. 5: 523. https://doi.org/10.3390/jof9050523