

Development of Glycerosomal pH Triggered In Situ Gelling System to Ameliorate the Nasal Delivery of Sulpiride for Pediatric Psychosis

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Statistical Design of Sulpiride-Loaded Glycerosomes
2.1.1. Box–Behnken Design (BBD) Analysis
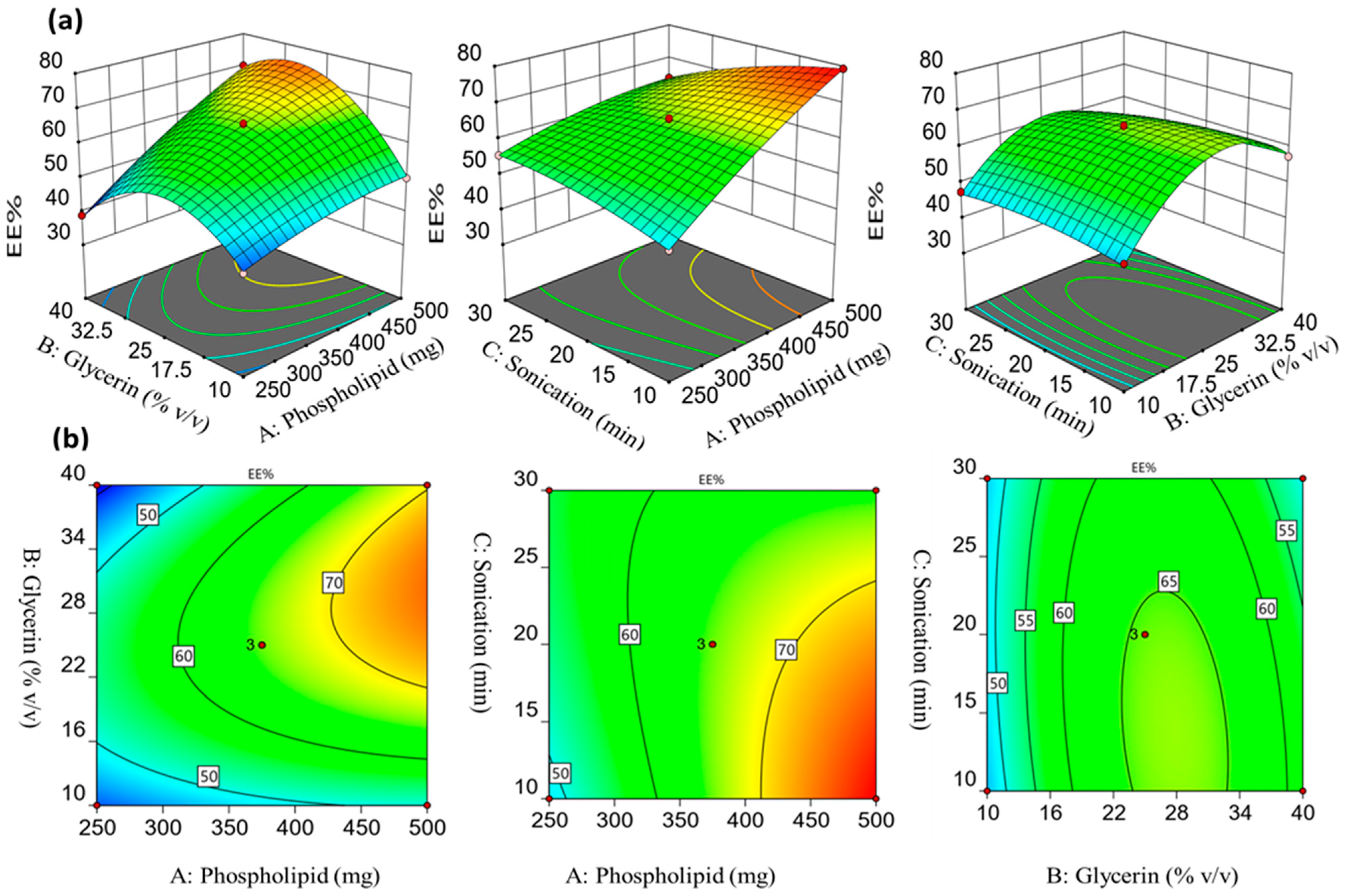
2.1.2. Influence of the Formulation Factors on the Encapsulation Efficiency (Y1)
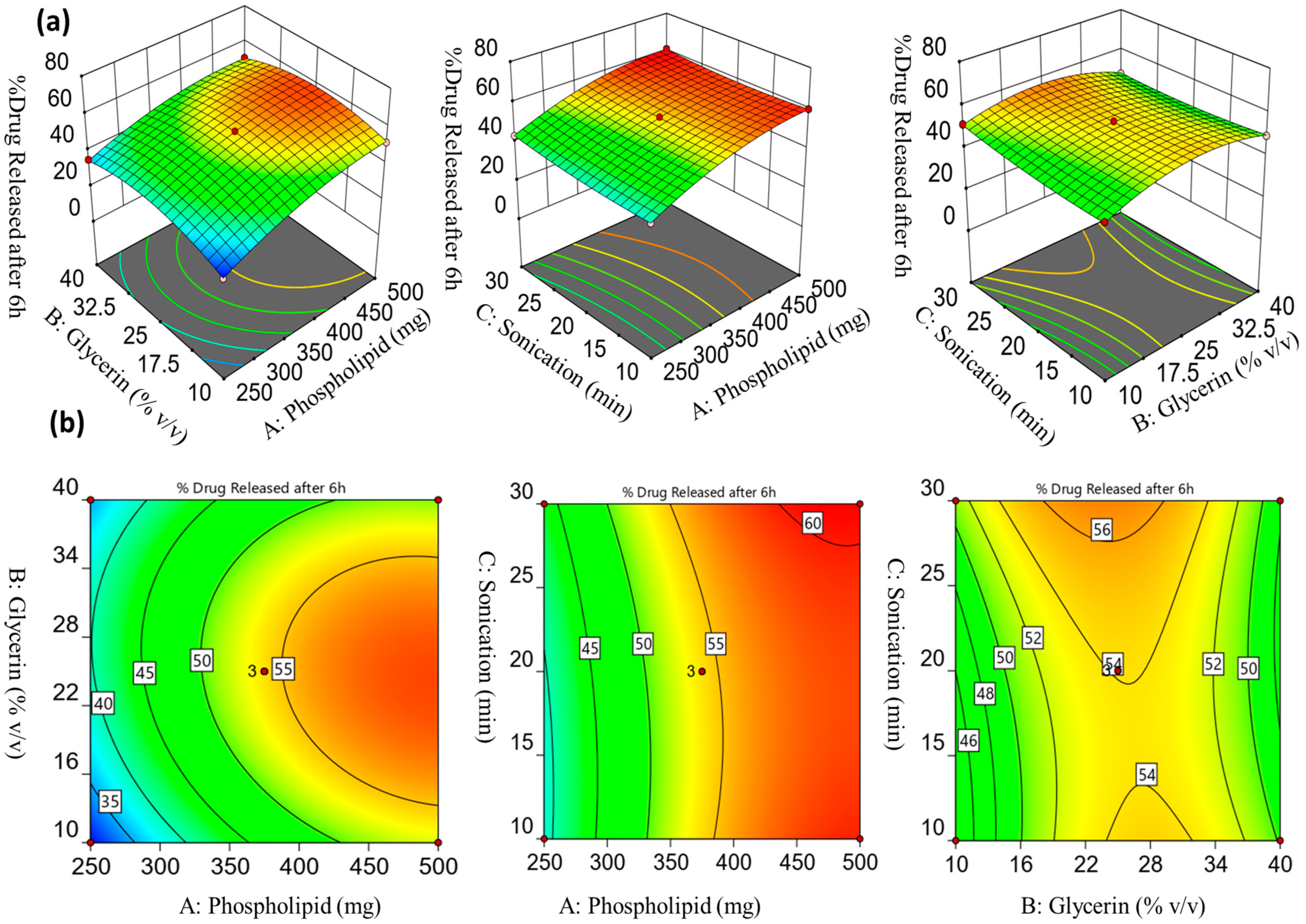
2.1.3. Influence of the Formulation Factors on the Drug Release % after 6 h (Y2)
2.1.4. Validation and Optimization Technique
2.2. Evaluation of the Optimized Sulpiride-Loaded Glycerosomal Formulation
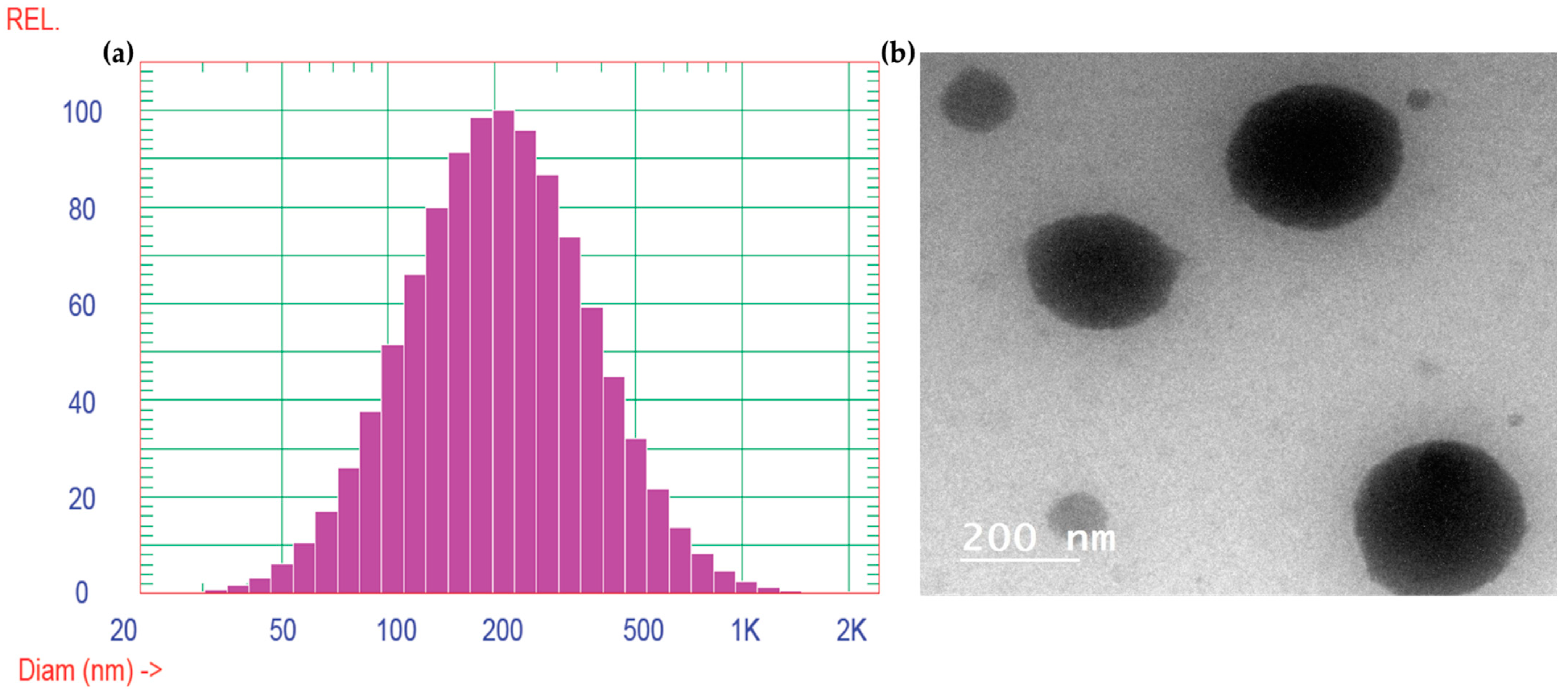
2.2.1. Vesicle Size, Zeta Potential Determination, and TEM Analysis
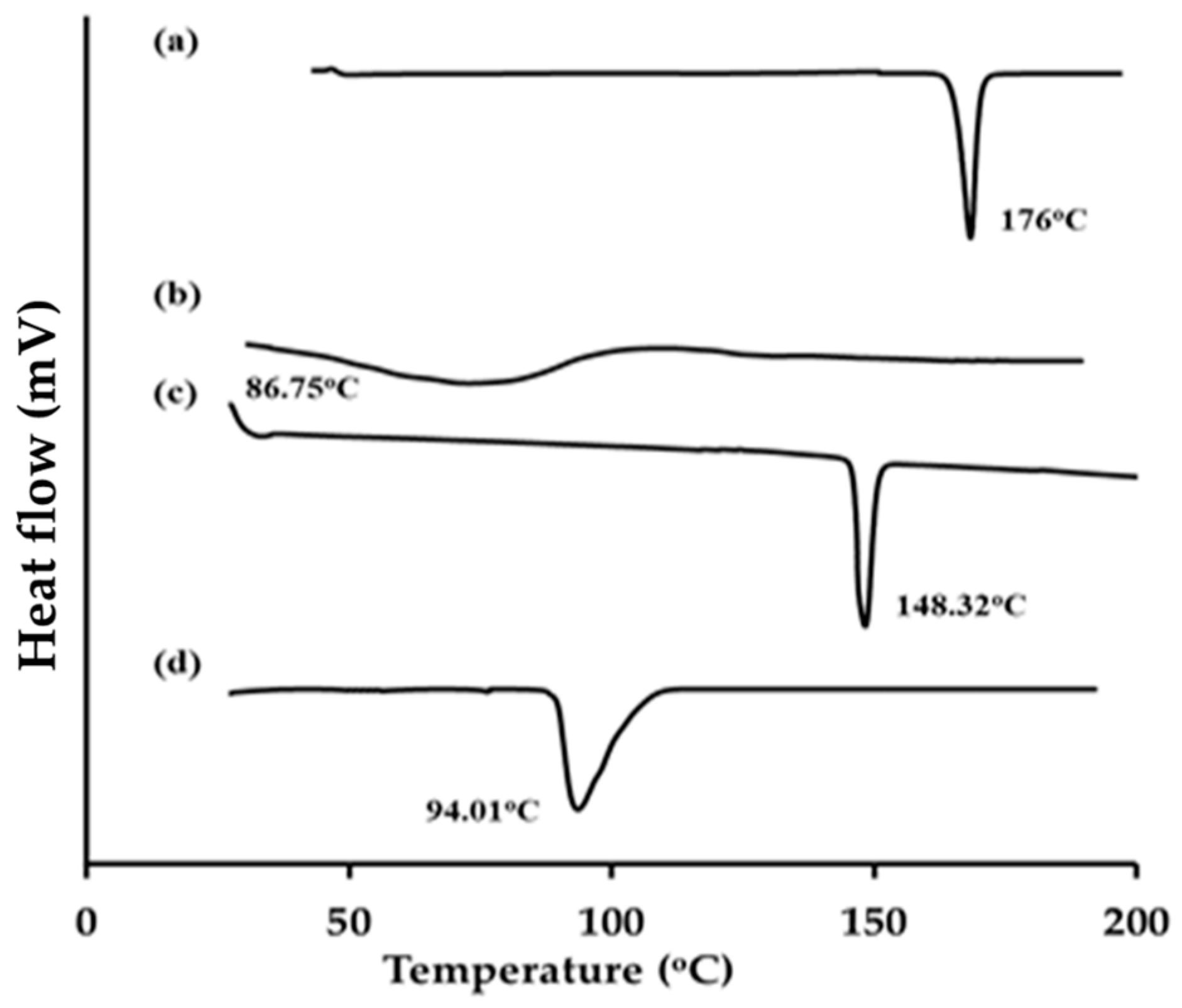
2.2.2. Differential Scanning Calorimetry
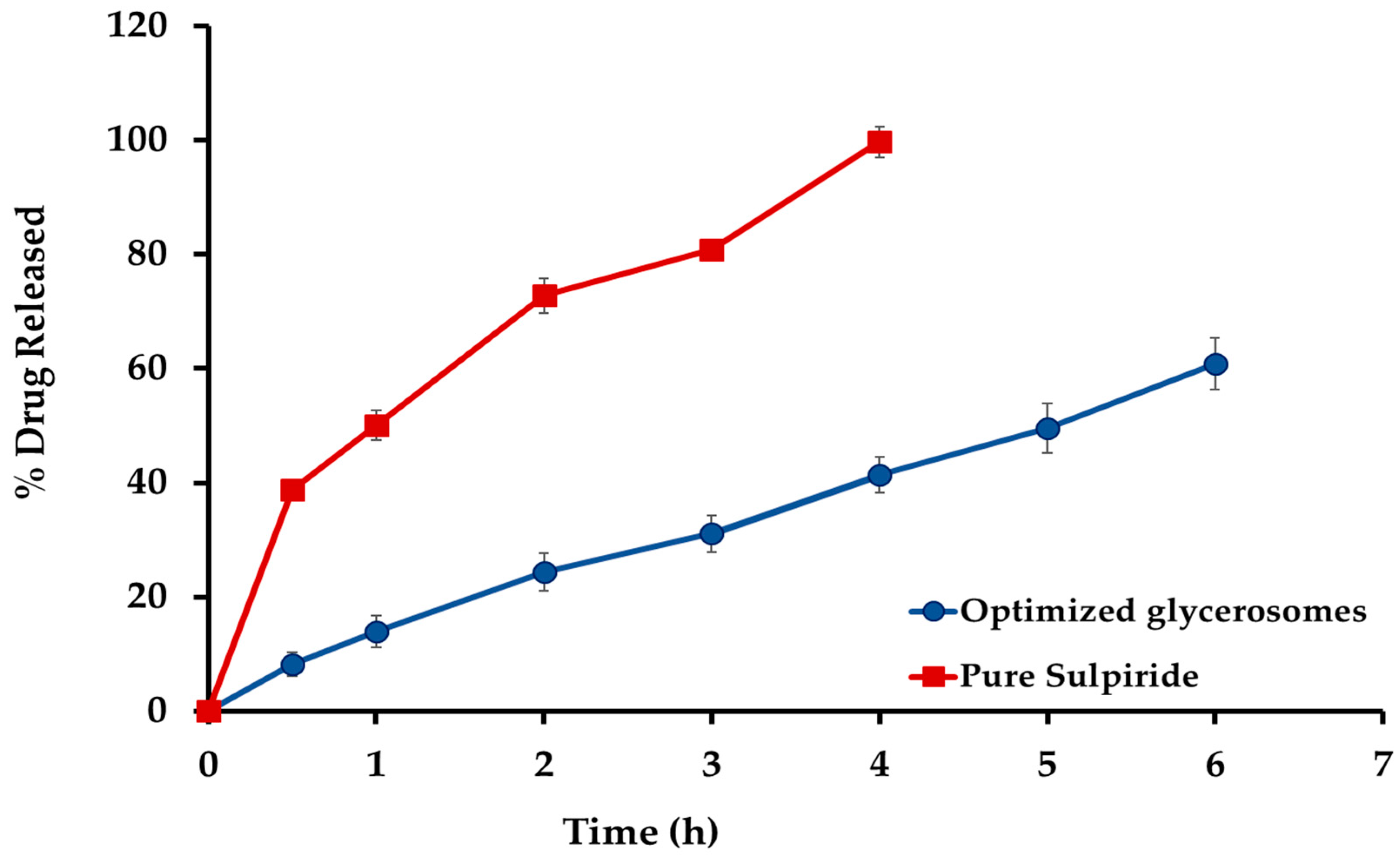
2.3. In Vitro Release Investigation
2.4. Evaluation of pH-Induced Carbopol In Situ Glycerosomal Gel
2.4.1. Physicochemical Evaluation of Carbopol Formulations
2.4.2. In Vitro Gelation and Viscosity of pH-Induced Carbopol Glycerosomal Gel In Situ Gels
2.4.3. Investigation of the Mucoadhesive Strength
2.4.4. Assessment of Spreadability
2.5. In Vitro Drug Release Studies Sul-GMs In Situ Gel Formulation
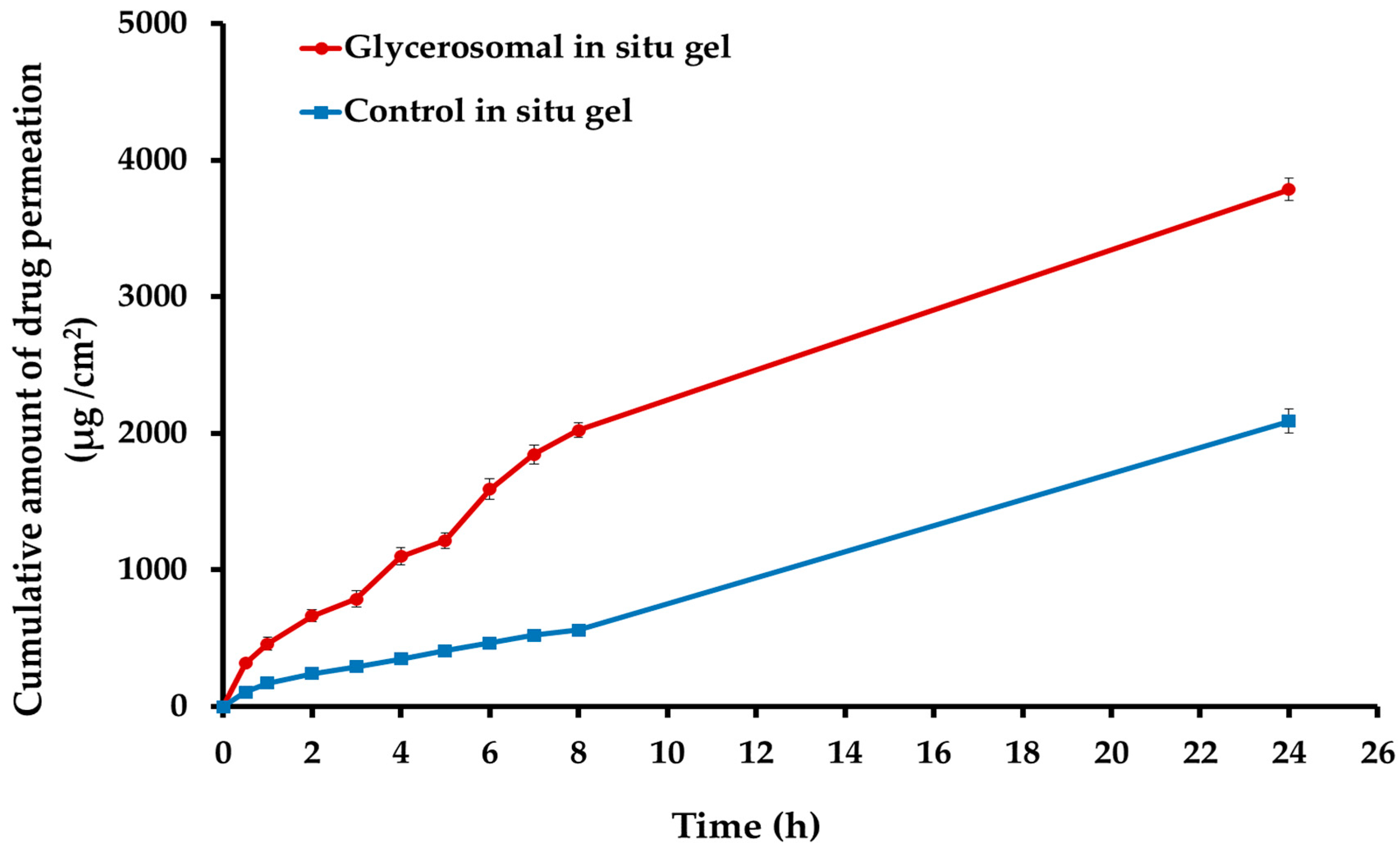
2.6. Ex Vivo Permeability Study of the Optimized Sulipride-Loaded Glycerosomal In Situ Gel
2.7. In Vivo Pharmacokinetic Study of the Optimized Sul-GMs
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Box-Behnken Design of Sulpiride-Loaded Glycerosomes
4.3. Manufacturing of Sulpiride Glycerosomes
4.4. Evaluation of Sulpiride-Loaded Glycerosomal Formulations
4.4.1. Entrapment Efficiency %
4.4.2. In Vitro Release Study of Sulpiride from the Generated Glycerosomal Preparations
4.5. Evaluation of the Optimum Sulpiride Formula
4.5.1. Vesicles’ Size (VS) and Zeta Potential (ZP)
4.5.2. Transmission Electron Microscopy (TEM)
4.5.3. Differential Scanning Calorimetry (DSC)
4.6. Manufacturing of pH-Induced Carbopol Glycerosomal In Situ Gel
4.7. Characterization of pH-Induced Carbopol Glycerosomal In Situ Gel
4.7.1. pH Value and Organoleptic Studies
4.7.2. Viscosity Determination
4.7.3. In Vitro Gelation Study and Gelation Capacity Determination
4.7.4. Gel Strength Assessment
4.7.5. Mucoadhesiveness Determination
4.7.6. Assessment of Spreadability
4.8. Determination of the In Vitro Drug Release of Glycerosomal In Situ Gel
4.9. Ex Vivo Drug Permeation Studies
4.10. Bioavailability Study of the Optimized Glycerosomal In Situ Gel
4.10.1. Study Design
4.10.2. HPLC Assay of Sulpiride
4.10.3. Pharmacokinetic Parameters
4.11. Analysis of Statistical Data
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Zidan, A.S.; Emam, S.E.; Shehata, T.M.; Ghazy, F.-e.S. Pediatric suppositories of sulpiride solid dispersion for treatment of Tourette syndrome: In vitro and in vivo investigations. AAPS PharmSciTech 2015, 16, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Birolo, R.; Bravetti, F.; Bordignon, S.; D’Abbrunzo, I.; Mazzeo, P.P.; Perissutti, B.; Bacchi, A.; Chierotti, M.R.; Gobetto, R. Overcoming the Drawbacks of Sulpiride by Means of New Crystal Forms. Pharmaceutics 2022, 14, 1754. [Google Scholar] [CrossRef]
- Kohri, N.; Naasani, I.; Iseki, K.; Miyazaki, K. Improving the oral bioavailability of sulpiride by a gastric-retained form in rabbits. J. Pharm. Pharmacol. 1996, 48, 371–374. [Google Scholar] [CrossRef]
- Ayoub, A.M.; Ibrahim, M.M.; Abdallah, M.H.; Mahdy, M.A. Sulpiride microemulsions as antipsychotic nasal drug delivery systems: In-vitro and pharmacodynamic study. J. Drug Deliv. Sci. Technol. 2016, 36, 10–22. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Ayoub, A.M.; Mahdy, M.A.E.; Abdallah, M.H. Solid Lipid Nanoparticles of Sulpiride: Improvement of Pharmacokinetic Properties. Int. J. Pharm. Investig. 2019, 9, 122–127. [Google Scholar] [CrossRef]
- Mohyeldin, S.M.; Samy, W.M.; Ragab, D.; Abdelmonsif, D.A.; Aly, R.G.; Elgindy, N.A. Precisely Fabricated Sulpiride-Loaded Nanolipospheres with Ameliorated Oral Bioavailability and Antidepressant Activity. Int. J. Nanomed. 2021, 16, 2013–2044. [Google Scholar] [CrossRef]
- Younes, N.F.; Habib, B.A. Augmented local skin accumulation efficiency of sertaconazole nitrate via glycerosomal hydrogel: Formulation, statistical optimization, ex vivo performance and in vivo penetration. J. Drug Deliv. Sci. Technol. 2022, 72, 103364. [Google Scholar] [CrossRef]
- Naguib, M.J.; Salah, S.; Abdel Halim, S.A.; Badr-Eldin, S.M. Investigating the potential of utilizing glycerosomes as a novel vesicular platform for enhancing intranasal delivery of lacidipine. Int. J. Pharm. 2020, 582, 119302. [Google Scholar] [CrossRef]
- Salem, H.F.; Kharshoum, R.M.; Sayed, O.M.; Abdel Hakim, L.F. Formulation design and optimization of novel soft glycerosomes for enhanced topical delivery of celecoxib and cupferron by Box–Behnken statistical design. Drug Dev. Ind. Pharm. 2018, 44, 1871–1884. [Google Scholar] [CrossRef]
- Melis, V.; Manca, M.L.; Bullita, E.; Tamburini, E.; Castangia, I.; Cardia, M.C.; Valenti, D.; Fadda, A.M.; Peris, J.E.; Manconi, M. Inhalable polymer-glycerosomes as safe and effective carriers for rifampicin delivery to the lungs. Colloids Surf. B Biointerfaces 2016, 143, 301–308. [Google Scholar] [CrossRef]
- Batchelor, H.K.; Marriott, J.F. Formulations for children: Problems and solutions. Br. J. Clin. Pharmacol. 2015, 79, 405–418. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Lee, Y.-B. Drug delivery to the brain via the nasal route of administration: Exploration of key targets and major consideration factors. J. Pharm. Investig. 2023, 53, 119–152. [Google Scholar] [CrossRef]
- Kushwaha, S.K.; Keshari, R.K.; Rai, A. Advances in nasal trans-mucosal drug delivery. J. Appl. Pharm. Sci. 2011, 1, 21–28. [Google Scholar]
- Nguyen, T.-T.-L.; Maeng, H.-J. Pharmacokinetics and pharmacodynamics of intranasal solid lipid nanoparticles and nanostructured lipid carriers for nose-to-brain delivery. Pharmaceutics 2022, 14, 572. [Google Scholar] [CrossRef]
- Singh, S.K.; Dadhania, P.; Vuddanda, P.R.; Jain, A.; Velaga, S.; Singh, S. Intranasal delivery of asenapine loaded nanostructured lipid carriers: Formulation, characterization, pharmacokinetic and behavioural assessment. RSC Adv. 2016, 6, 2032–2045. [Google Scholar] [CrossRef]
- Bhanderi, M.; Shah, J.; Gorain, B.; Nair, A.B.; Jacob, S.; Asdaq, S.M.B.; Fattepur, S.; Alamri, A.S.; Alsanie, W.F.; Alhomrani, M. Optimized rivastigmine nanoparticles coated with eudragit for intranasal application to brain delivery: Evaluation and nasal ciliotoxicity studies. Materials 2021, 14, 6291. [Google Scholar] [CrossRef]
- Duong, V.-A.; Nguyen, T.-T.-L.; Maeng, H.-J. Recent advances in intranasal liposomes for drug, gene, and vaccine delivery. Pharmaceutics 2023, 15, 207. [Google Scholar] [CrossRef]
- Hamzah, M.L.; Kassab, H.J. Formulation and Characterization of Intranasal Drug Delivery of Frovatriptan-Loaded Binary Ethosomes Gel for Brain Targeting. Nanotechnol. Sci. Appl. 2024, 17, 1–19. [Google Scholar] [CrossRef]
- Kaur, A.; Nigam, K.; Srivastava, S.; Tyagi, A.; Dang, S. Memantine nanoemulsion: A new approach to treat Alzheimer’s disease. J. Microencapsul. 2020, 37, 355–365. [Google Scholar] [CrossRef]
- Singh, K.; HariKumar, S. Injectable in-situ gelling controlled release drug delivery system. Int. J. Drug Dev. Res. 2012, 4, 56–69. [Google Scholar]
- Kurniawansyah, I.S.; Rusdiana, T.; Sopyan, I.; Desy Arya, I.F.; Wahab, H.A.; Nurzanah, D. Comparative Study of In Situ Gel Formulation Based on the Physico-Chemical Aspect: Systematic Review. Gels 2023, 9, 645. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.; Li, Z.; Li, N.; Feng, N. Essential oil-mediated glycerosomes increase transdermal paeoniflorin delivery: Optimization, characterization, and evaluation in vitro and in vivo. Int. J. Nanomed. 2017, 12, 3521–3532. [Google Scholar] [CrossRef]
- Abdallah, M.H.; Elghamry, H.A.; Khalifa, N.E.; Khojali, W.M.; Khafagy, E.-S.; Shawky, S.; El-Horany, H.E.-S.; El-Housiny, S. Development and Optimization of Erythromycin Loaded Transethosomes Cinnamon Oil Based Emulgel for Antimicrobial Efficiency. Gels 2023, 9, 137. [Google Scholar] [CrossRef]
- Abdallah, M.H.; Elghamry, H.A.; Khalifa, N.E.; Khojali, W.M.A.; Khafagy, E.-S.; Lila, A.S.A.; El-Horany, H.E.-S.; El-Housiny, S. Ginger Extract-Loaded Sesame Oil-Based Niosomal Emulgel: Quality by Design to Ameliorate Anti-Inflammatory Activity. Gels 2022, 8, 737. [Google Scholar] [CrossRef]
- Abdallah, M.H.; Abu Lila, A.S.; Shawky, S.M.; Almansour, K.; Alshammari, F.; Khafagy, E.-S.; Makram, T.S. Experimental Design and Optimization of Nano-Transfersomal Gel to Enhance the Hypoglycemic Activity of Silymarin. Polymers 2022, 14, 508. [Google Scholar] [CrossRef]
- Said, M.; Aboelwafa, A.A.; Elshafeey, A.H.; Elsayed, I. Central composite optimization of ocular mucoadhesive cubosomes for enhanced bioavailability and controlled delivery of voriconazole. J. Drug Deliv. Sci. Technol. 2021, 61, 102075. [Google Scholar] [CrossRef]
- Zaki, R.M.; Alfadhel, M.M.; Alossaimi, M.A.; Elsawaf, L.A.; Devanathadesikan Seshadri, V.; Almurshedi, A.S.; Yusif, R.M.; Said, M. Central Composite Optimization of Glycerosomes for the Enhanced Oral Bioavailability and Brain Delivery of Quetiapine Fumarate. Pharmaceuticals 2022, 15, 940. [Google Scholar] [CrossRef]
- Jahanfar, S.; Gahavami, M.; Khosravi-Darani, K.; Jahadi, M.; Mozafari, M.R. Entrapment of rosemary extract by liposomes formulated by Mozafari method: Physicochemical characterization and optimization. Heliyon 2021, 7, e08632. [Google Scholar] [CrossRef]
- Naeem, M.; Rahman, N.U.; TAVARES, G.; Barbosa, S.F.; Chacra, N.B.; Loebenberg, R.; Sarfraz, M.K. Physicochemical, in vitro and in vivo evaluation of flurbiprofen microemulsion. An. Acad. Bras. Ciências 2015, 87, 1823–1831. [Google Scholar] [CrossRef]
- Gaba, B.; Khan, T.; Haider, M.F.; Alam, T.; Baboota, S.; Parvez, S.; Ali, J. Vitamin E loaded naringenin nanoemulsion via intranasal delivery for the management of oxidative stress in a 6-OHDA Parkinson’s disease model. BioMed Res. Int. 2019, 2019, 2382563. [Google Scholar] [CrossRef] [PubMed]
- Bharath, S.; Karuppaiah, A.; Siram, K.; Hariharan, S.; Santhanam, R.; Veintramuthu, S. Development and evaluation of a pH triggered in situ ocular gel of brimonidine tartrate. J. Res. Pharm. 2020, 24, 416–424. [Google Scholar] [CrossRef]
- Mahathi, K. Formulation and Evaluation of Nasal In Situ Gel of Levofloxacin Hemihydrate. Indo Am. J. Pharm. Res. 2014, 4, 5817–5827. [Google Scholar]
- Satyananda, S. pH-induced in situ gelling system of an anti-infective drug for sustained ocular delivery. J. Appl. Pharm. Sci. 2014, 4, 101–104. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, S.; Wang, H.; Bie, H. A mucoadhesive, thermoreversible in situ nasal gel of geniposide for neurodegenerative diseases. PLoS ONE 2017, 12, e0189478. [Google Scholar] [CrossRef]
- Chaudhary, B.; Verma, S. Preparation and evaluation of novel in situ gels containing acyclovir for the treatment of oral herpes simplex virus infections. Sci. World J. 2014, 2014, 280928. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.H.; Lila, A.S.A.; Anwer, M.K.; Khafagy, E.-S.; Mohammad, M.; S Soliman, M. Formulation, development and evaluation of ibuprofen loaded nano-transferosomal gel for the treatment of psoriasis. J. Pharm. Res. Int. 2019, 31, 1–8. [Google Scholar] [CrossRef]
- Russo, E.; Villa, C. Poloxamer hydrogels for biomedical applications. Pharmaceutics 2019, 11, 671. [Google Scholar] [CrossRef]
- Idrees, M.; Rahman, N.; Ahmad, S.; Ali, M.; Ahmad, I. Enhance transdermal delivery of flurbiprofen via microemulsions: Effects of different types of surfactants and cosurfactants. DARU J. Pharm. Sci. 2011, 19, 433. [Google Scholar]
- Zhu, C.; Zhang, Y.; Wu, T.; He, Z.; Guo, T.; Feng, N. Optimizing glycerosome formulations via an orthogonal experimental design to enhance transdermal triptolide delivery. Acta Pharm. 2022, 72, 135–146. [Google Scholar] [CrossRef]
- Md, S.; Alhakamy, N.A.; Aldawsari, H.M.; Husain, M.; Khan, N.; Alfaleh, M.A.; Asfour, H.Z.; Riadi, Y.; Bilgrami, A.L.; Akhter, M.H. Plumbagin-Loaded Glycerosome Gel as Topical Delivery System for Skin Cancer Therapy. Polymers 2021, 13, 923. [Google Scholar] [CrossRef] [PubMed]
- Said, M.; Elmenoufy, G.A. Evaluation of quetiapine fumarate and its solid lipid nanoparticles as antipsychotic drug in rat model of schizophrenia. Biomed. Res. Ther. 2017, 4, 1480–1497. [Google Scholar]
- Abdallah, M.H.; Shahien, M.M.; Alshammari, A.; Ibrahim, S.; Ahmed, E.H.; Atia, H.A.; Elariny, H.A. The Exploitation of Sodium Deoxycholate-Stabilized Nano-Vesicular Gel for Ameliorating the Antipsychotic Efficiency of Sulpiride. Gels 2024, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Moolakkadath, T.; Aqil, M.; Ahad, A.; Imam, S.S.; Praveen, A.; Sultana, Y.; Mujeeb, M. Preparation and optimization of fisetin loaded glycerol based soft nanovesicles by Box-Behnken design. Int. J. Pharm. 2020, 578, 119125. [Google Scholar] [CrossRef]
- Abdelnabi, D.M.; Abdallah, M.H.; Elghamry, H.A. Buspirone hydrochloride loaded in situ nanovesicular gel as an anxiolytic nasal drug delivery system: In vitro and animal studies. AAPS PharmSciTech 2019, 20, 134. [Google Scholar] [CrossRef]
- Hosny, K.M.; Rizg, W.Y.; Khallaf, R.A. Preparation and Optimization of In Situ Gel Loaded with Rosuvastatin-Ellagic Acid Nanotransfersomes to Enhance the Anti-Proliferative Activity. Pharmaceutics 2020, 12, 263. [Google Scholar] [CrossRef]
- Abdallah, M.H.; Abdelnabi, D.M.; Elghamry, H.A. Response Surface Methodology for Optimization of Buspirone Hydrochloride-Loaded In Situ Gel for Pediatric Anxiety. Gels 2022, 8, 395. [Google Scholar] [CrossRef]
- Sherafudeen, S.P.; Vasantha, P.V. Development and evaluation of in situ nasal gel formulations of loratadine. Res. Pharm. Sci. 2015, 10, 466–476. [Google Scholar]
- Bayan, M.F.; Chandrasekaran, B.; Alyami, M.H. Development and Characterization of Econazole Topical Gel. Gels 2023, 9, 929. [Google Scholar] [CrossRef]
- Salunke, S.R.; Patil, S.B. Ion activated in situ gel of gellan gum containing salbutamol sulphate for nasal administration. Int. J. Biol. Macromol. 2016, 87, 41–47. [Google Scholar] [CrossRef]
- Sanjana, A.; Ahmed, M.G.; Bh, J.G. Preparation and evaluation of in-situ gels containing hydrocortisone for the treatment of aphthous ulcer. J. Oral Biol. Craniofacial Res. 2021, 11, 269–276. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Independent Variables | Dependent Variables | |||
|---|---|---|---|---|---|
| A (mg) | B (%) | C (min) | Y1 (%) | Y2 (%) | |
| F1 | 375 | 40 | 10 | 57.43 ± 1.09 | 49.41 ± 2.78 |
| F2 | 500 | 25 | 10 | 79.32 ± 1.00 | 59.97 ± 2.38 |
| F3 | 500 | 25 | 30 | 63.98 ± 0.99 | 60.94 ± 0.86 |
| F4 | 250 | 25 | 10 | 47.98 ± 0.50 | 38.74 ± 0.73 |
| F5 | 250 | 40 | 20 | 38.87 ± 0.71 | 35.38 ± 1.63 |
| F6 | 250 | 25 | 30 | 55.60 ± 1.02 | 43.36 ± 2.21 |
| F7 | 375 | 25 | 20 | 65.96 ± 1.91 | 54.02 ± 1.35 |
| F8 | 375 | 40 | 30 | 49.18 ± 1.89 | 47.94 ±1.46 |
| F9 | 250 | 10 | 20 | 41.81 ± 0.67 | 29.74 ± 2.22 |
| F10 | 500 | 40 | 20 | 70.07 ± 0.69 | 50.91 ± 1.97 |
| F11 | 375 | 25 | 20 | 64.92 ± 2.09 | 53.74 ± 0.92 |
| F12 | 375 | 25 | 20 | 65.63 ± 1.20 | 54.44 ± 0.76 |
| F13 | 375 | 10 | 10 | 46.78 ± 2.52 | 44.61 ± 1.04 |
| F14 | 375 | 10 | 30 | 47.51 ± 1.87 | 51.33 ± 0.53 |
| F15 | 500 | 10 | 20 | 49.98 ± 0.65 | 51.80 ± 0.75 |
| Formulation | Gel Base | HPMC Concentration (% w/w) | Gelation Capacity | pH | Spreadability (cm) | Gel Strength (s) | Mucoadhesive Strength (dyne/cm2) | Viscosity (cP) |
|---|---|---|---|---|---|---|---|---|
| G1 | Carbopol 974P (0.2% w/w) | 0.2 | - | ND | ND | ND | ND | ND |
| 0.4 | + | 6.36 ± 0.17 | 4.62 ± 0.08 | 14.37 ± 0.78 | 1949.43 ± 14.64 | 5848.30 ± 43.93 | ||
| 0.6 | + | 6.48 ± 0.09 | 4.38 ± 0.11 | 19.67 ± 0.86 | 2365.57 ± 20.05 | 6096.70 ± 60.15 | ||
| 0.8 | ++ | 5.79 ± 0.19 | 4.22 ± 0.58 | 23.57 ± 0.95 | 2572.77 ± 29.81 | 6784.97 ± 38.48 | ||
| 1.0 | +++ | 6.30 ± 0.15 | 4.02 ± 0.13 | 27.80 ± 0.26 | 2723.87 ± 27.54 | 7204.93 ± 64.79 | ||
| G2 | Carbopol 974P (0.4% w/w) | 0.2 | - | ND | ND | ND | ND | ND |
| 0.4 | + | 6.42 ± 0.19 | 4.42 ± 0.08 | 12.67 ± 0.76 | 2721.20 ± 14.70 | 7163.6 ± 44.10 | ||
| 0.6 | ++ | 6.49 ± 0.17 | 4.25 ± 0.10 | 19.37 ± 0.91 | 2889.47 ± 24.60 | 7725.17 ± 73.81 | ||
| 0.8 | +++ | 6.07 ± 0.14 | 4.08 ± 0.12 | 35.27 ± 0.70 | 3030.23 ± 18.74 | 8090.70 ± 56.22 | ||
| 1.0 | +++ | 5.89 ± 0.08 | 3.82 ± 0.10 | 38.50 ± 0.83 | 3282.73 ± 10.90 | 8848.20 ± 32.71 | ||
| G3 | Carbopol 974P (0.6% w/w) | 0.2 | + | 5.79 ±0.19 | 3.93 ± 0.15 | 11.53 ± 0.45 | 3188.24 ± 16.49 | 8711.73 ± 36.39 |
| 0.4 | ++ | 6.30 ± 0.15 | 3.68 ± 0.08 | 21.77 ± 0.49 | 3549.23 ± 14.93 | 9090.70 ± 56.22 | ||
| 0.6 | ++ | 6.38 ± 0.13 | 3.47 ± 0.06 | 31.03 ± 0.55 | 3927.50 ± 33.21 | 9848.20 ± 32.70 | ||
| 0.8 | +++ | 6.40 ± 0.09 | 3.22 ± 0.10 | 42.03 ± 0.45 | 4175.90 ± 27.93 | 10,046.40 ± 34.48 | ||
| 1.0 | +++ | 6.07 ±0.12 | 3.02 ± 0.13 | 52.13 ± 0.32 | 4682.133 ± 11.49 | 10,148.07 ± 36.30 |
| Parameters | Oral Marketed Product | Control Sul Gel | Optimized Sul-GMs Gel |
|---|---|---|---|
| Cmax (ng/mL) | 698.56 ± 42.06 | 248.87 ± 27.46 | 500.26 ± 32.01 |
| tmax (h) | 2 | 3 | 6 |
| Kel (h−1) | 0.13 ± 0.01 | 0.07± 0.01 | 0.062 ± 0.02 |
| t1/2 (h) | 5.41 ± 046 | 9.63 ± 1.34 | 12.04 ± 3.7 |
| AUC0–t (ng/mL−1·h) | 2388.41 ± 68.04 | 1179.45 ± 169.02 | 4540.27 ± 295.48 |
| MRT (h) | 4.79 ± 0.11 | 11.28 ± 1.86 | 12.99 ± 3.97 |
| Relative bioavailability (%) | 202.50 | - | 384.85% |
| Independent Variables | Type | Actual Levels | |
|---|---|---|---|
| Low | High | ||
| A: Lipid amount (mg) | Numeric | 250 | 500 |
| B: Glycerin concentration v/v (%) | Numeric | 10 | 40 |
| C: Time of sonication (Minutes) | Numeric | 10 | 30 |
| Response Variables (Dependent Variables) | Goal | ||
| (Y1) = Encapsulation efficiency EE% | Maximize | ||
| (Y2) = In vitro release of the drug after 6 h | Maximize | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahien, M.M.; Alshammari, A.; Ibrahim, S.; Ahmed, E.H.; Atia, H.A.; Elariny, H.A.; Abdallah, M.H. Development of Glycerosomal pH Triggered In Situ Gelling System to Ameliorate the Nasal Delivery of Sulpiride for Pediatric Psychosis. Gels 2024, 10, 608. https://doi.org/10.3390/gels10090608
Shahien MM, Alshammari A, Ibrahim S, Ahmed EH, Atia HA, Elariny HA, Abdallah MH. Development of Glycerosomal pH Triggered In Situ Gelling System to Ameliorate the Nasal Delivery of Sulpiride for Pediatric Psychosis. Gels. 2024; 10(9):608. https://doi.org/10.3390/gels10090608
Chicago/Turabian StyleShahien, Mona M., Alia Alshammari, Somaia Ibrahim, Enas Haridy Ahmed, Hanan Abdelmawgoud Atia, Hemat A. Elariny, and Marwa H. Abdallah. 2024. "Development of Glycerosomal pH Triggered In Situ Gelling System to Ameliorate the Nasal Delivery of Sulpiride for Pediatric Psychosis" Gels 10, no. 9: 608. https://doi.org/10.3390/gels10090608
APA StyleShahien, M. M., Alshammari, A., Ibrahim, S., Ahmed, E. H., Atia, H. A., Elariny, H. A., & Abdallah, M. H. (2024). Development of Glycerosomal pH Triggered In Situ Gelling System to Ameliorate the Nasal Delivery of Sulpiride for Pediatric Psychosis. Gels, 10(9), 608. https://doi.org/10.3390/gels10090608