

HGSMDA: miRNA–Disease Association Prediction Based on HyperGCN and Sørensen-Dice Loss



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Information about miRNAs, information about diseases, and information about known miRNA–disease associations were integrated. By integrating this information into the HGSMDA model, we were able to more fully characterize the relationship between miRNAs and disease.
- HyperGCN was introduced to construct a miRNA–disease heterogeneous hypergraph using hypernodes, and GCNs were trained on the graph to aggregate information.
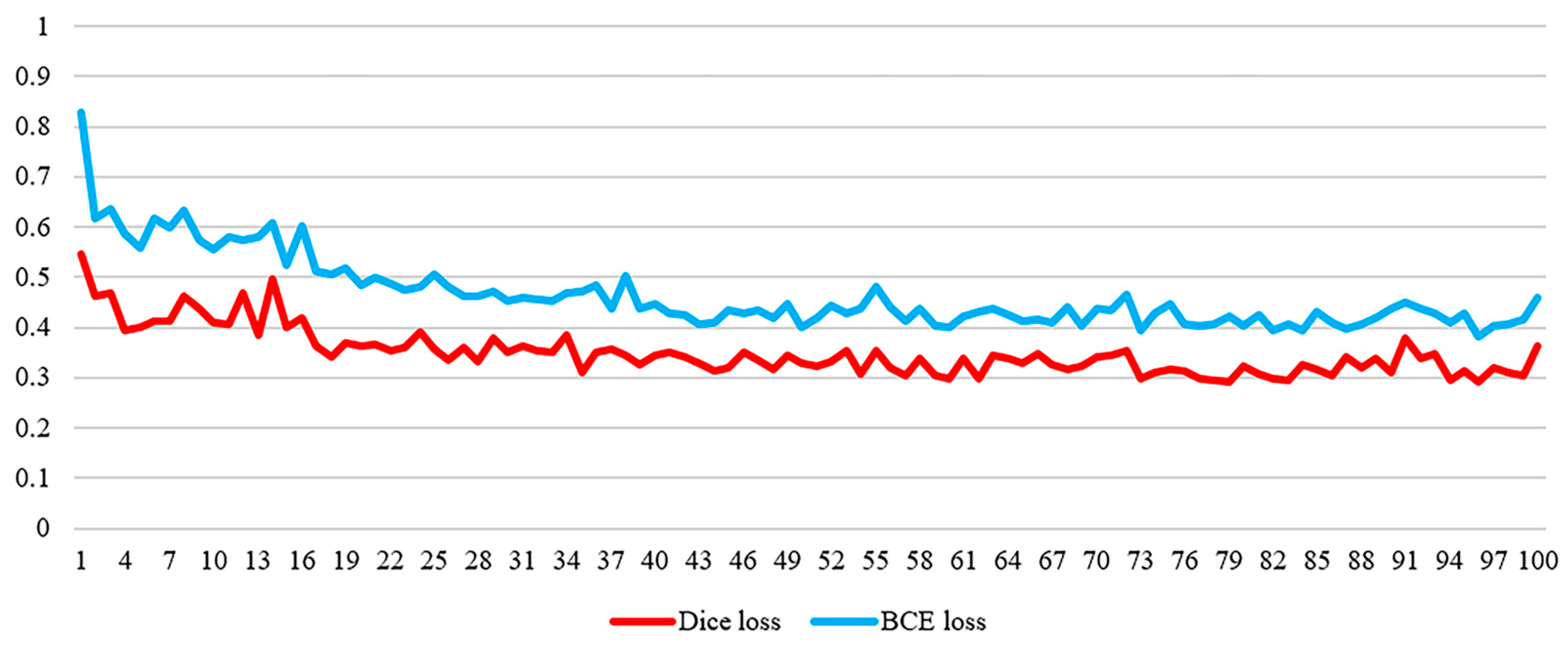
- The Sørensen-Dice loss function is employed to evaluate the likeness between the predicted outcomes and the actual values. This facilitates a more precise evaluation of the model’s capability.
2. Data and Experiments
2.1. Datasets
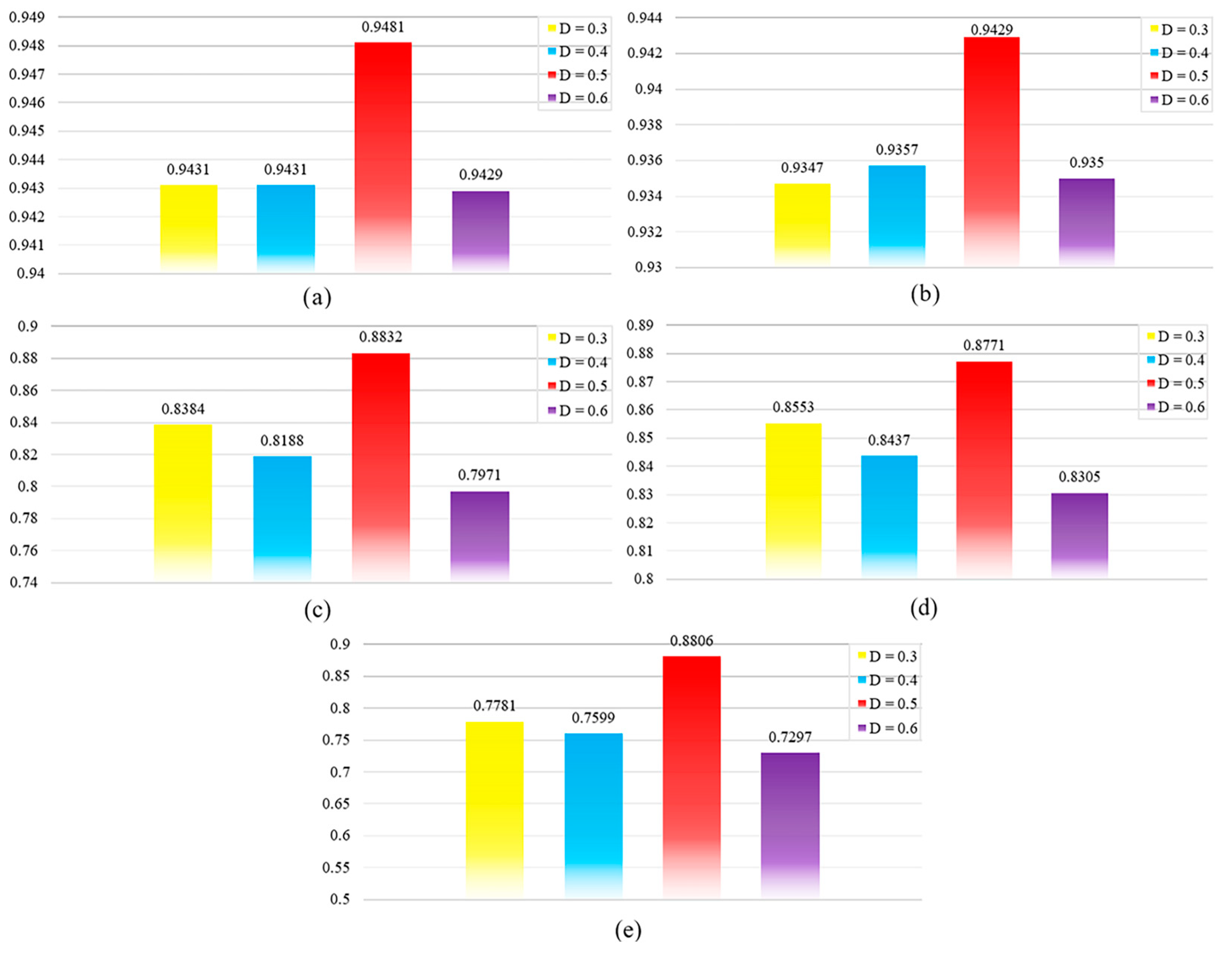
2.2. Parametric Analysis
2.2.1. Dropout Parameter Settings
2.2.2. Feature Embedding Dimension
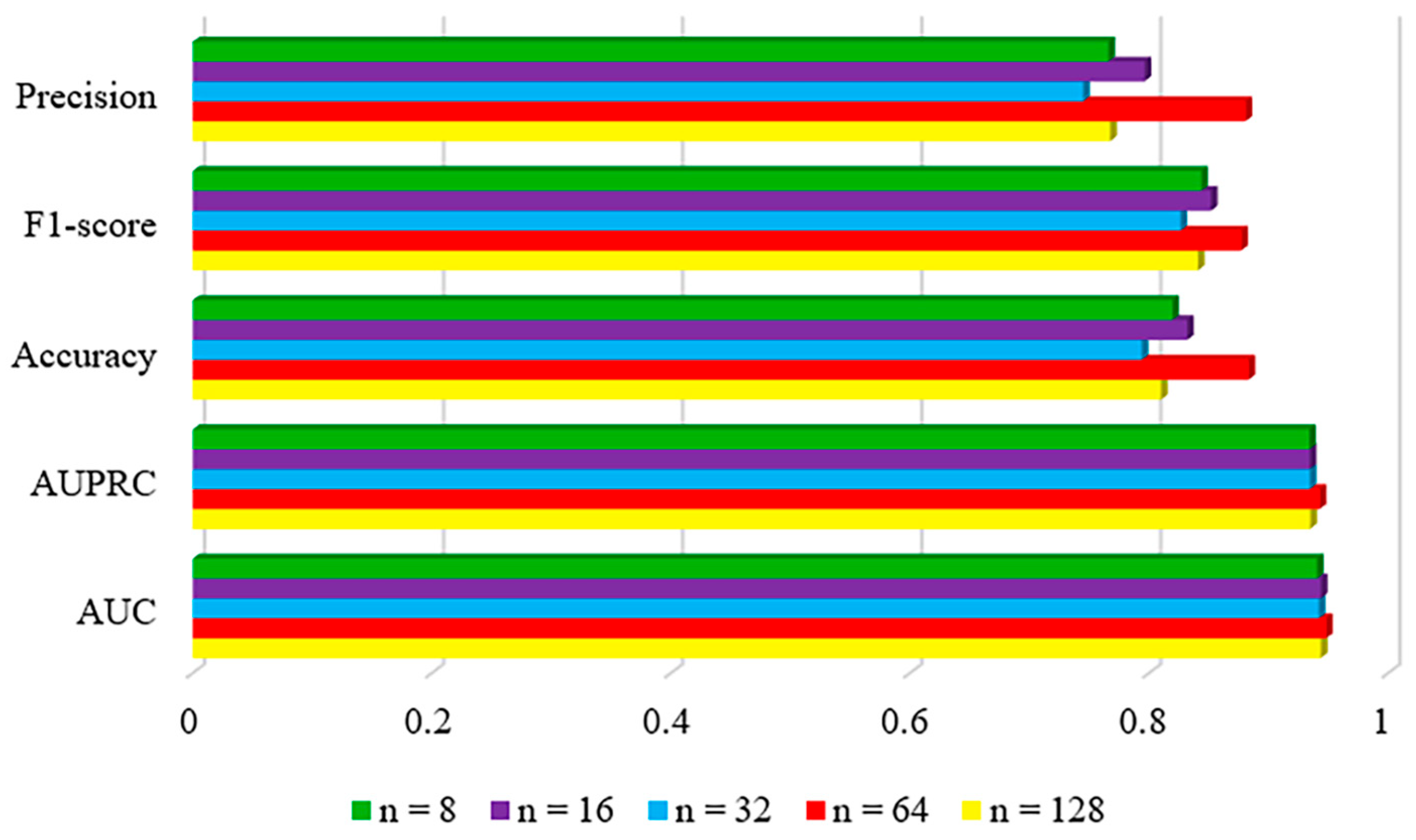
2.2.3. The Number of Hypernodes
3. Results and Discussion
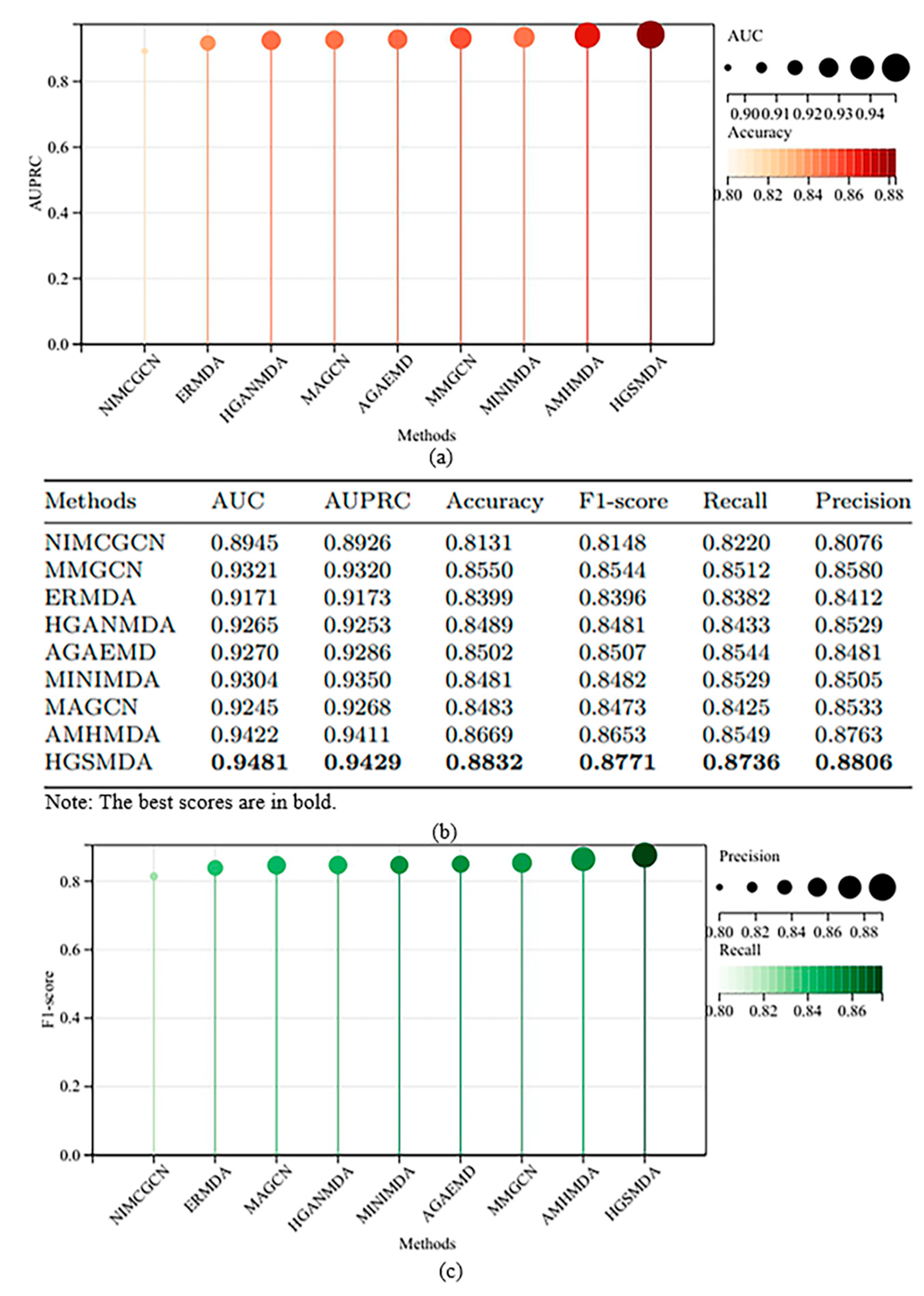
3.1. Comparison with Other Methods
- NIMCGCN [15] uses GCN to learn miRNA and disease potential characteristic representations. It inputs the learned characteristics into the NIMC model to produce a matrix of association complements to forecast miRNA–disease associations.
- MMGCN [16] uses a GCN encoder to obtain characteristics under different similarity views separately and forecasts miRNA–disease associations by utilizing multi-channel attention to enhance the learned potential representation of association prediction.
- ERMDA [22] proposed a resampling strategy to construct multiple subsets and applied feature selection methods to increase the diversity among these subsets. It then uses soft voting to forecast the connections of miRNAs with diseases.
- HGANMDA [23] constructs a heterogeneous graph, applies node-level attention to learn neighboring nodes, applies semantic-level attention to learn meta-paths, and lastly employs a bilinear decoder to reconstruct miRNA–disease associations.
- AGAEMD [24] creates heterogeneous matrices and uses autoencoders in miRNA–disease networks to polymerize information and reconstruct miRNA–disease association networks.
- MINIMDA [25] constructs disease similarity networks to obtain embedding representations by mixing higher-order neighborhood information, which is fed into a multilayer perceptron (MLP) to forecast potential connections between miRNAs and diseases.
- MAGCN [26] used lncRNA-miRNA interactions to predict novel miRNA–disease correlations through graph convolutional networks with attentional mechanisms and convolutional neural network combiners.
- AMHMDA [27] constructed multiple similarity networks, introduced virtual nodes to construct heterogeneous hypergraphs, and used the output of graph convolutional networks to predict associations.
3.2. Ablation Experiments
3.3. Case Study
4. Methodology
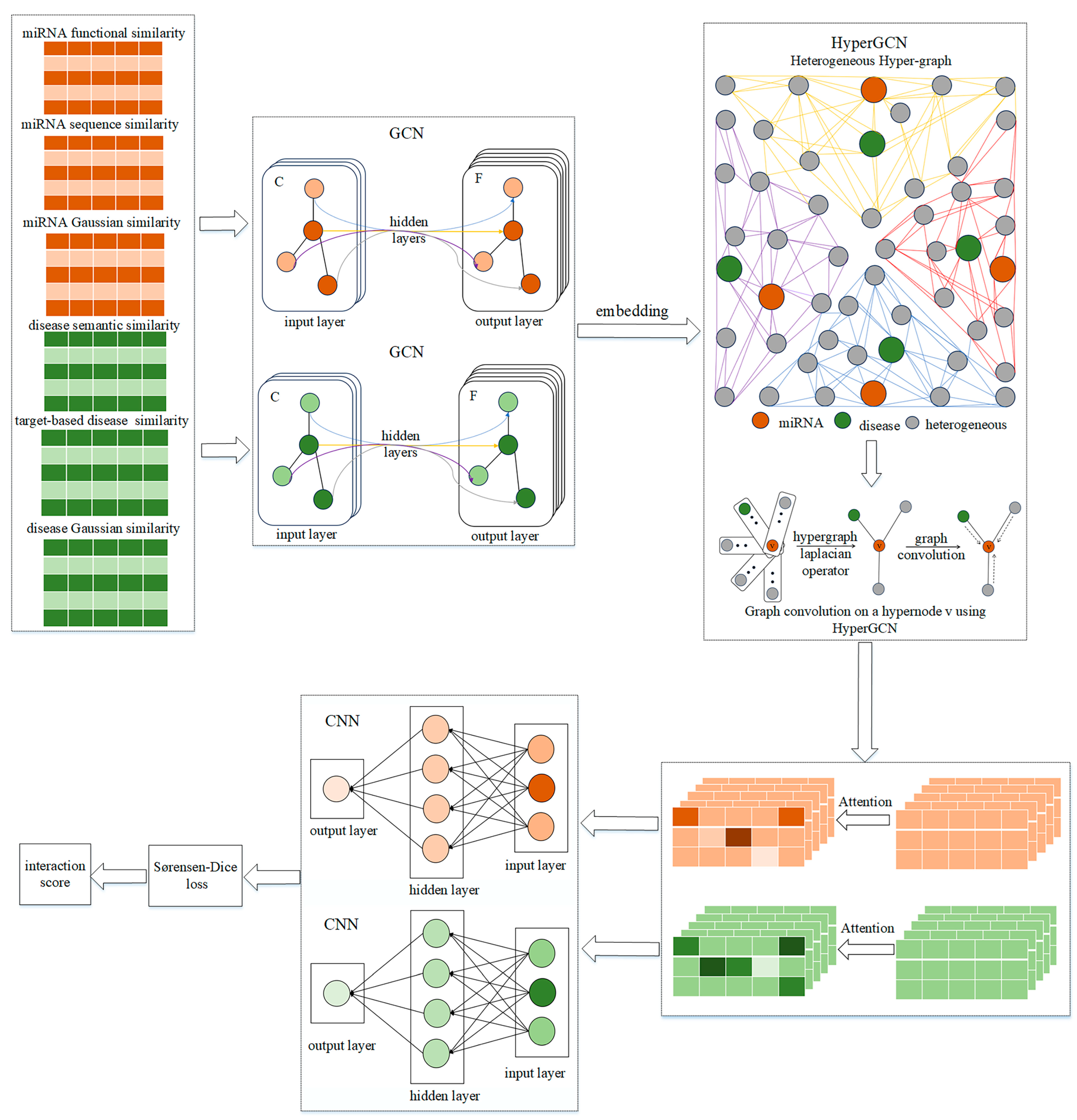
4.1. HGSMDA Framework
- Extracting features: We constructed multiple miRNA and disease similarity networks and used GCN for extracting information from different perspectives.
- HyperGCN: We introduce HyperGCN to construct a miRNA–disease heteromorphic hypergraph using hypernodes, and train GCN on the graph to aggregate information.
- Measuring the degree of similarity: We leverage the attention mechanism to fuse the output of the HyperGCN layer in combination with a CNN (Convolutional Neural Network) for classification. We then use the Sørensen-Dice loss function to scale the degree of similarity between the predictions and the true values.
4.2. Extraction of Features
4.3. HyperGCN
4.4. Measuring Similarity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.X.; Yan, G.Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Xuan, P.; Wang, X.; Zhang, T.; Li, J.; Liu, Y.; Zhang, W. A non-negative matrix factorization based method for predicting disease-associated miRNAs in miRNA-disease bilayer network. Bioinformatics 2018, 34, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA-Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.-X.; Lv, J.-Y.; Li, Y.-S.; Xiao, Y.; Shao, T.-T.; Huo, X.; Li, X.; Zou, Y.; Han, Q.-L.; et al. Prioritizing Candidate Disease miRNAs by Topological Features in the miRNA Target-Dysregulated Network: Case Study of Prostate Cancer. Mol. Cancer Ther. 2011, 10, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.N.; Li, J.Q. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 2019, 35, 4730–4738. [Google Scholar] [CrossRef]
- Shang, J.; Yang, Y.; Li, F.; Guan, B.; Liu, J.-X.; Sun, Y. BLNIMDA: Identifying miRNA-disease associations based on weighted bi-level network. BMC Genom. 2022, 23, 686. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, Y.; Valsdottir, L.R.; Zang, T.; Peng, J. Identifying drug-target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. 2021, 22, 2141–2150. [Google Scholar] [CrossRef]
- Li, Y.; Qiao, G.; Wang, K.; Wang, G. Drug-target interaction predication via multi-channel graph neural networks. Brief. Bioinform. 2022, 23, bbab346. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Tang, Q.; Dai, W.; Chen, T. Improving cancer driver gene identification using multi-task learning on graph convolutional network. Brief. Bioinform. 2022, 23, bbab432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Liu, T.; Ning, C.; Zhang, Z.; Zhou, W. Neural inductive matrix completion with graph convolutional networks for miRNA-disease association prediction. Bioinformatics 2020, 36, 2538–2546. [Google Scholar] [CrossRef]
- Tang, X.; Luo, J.; Shen, C.; Lai, Z. Multi-view Multichannel Attention Graph Convolutional Network for miRNA-disease association prediction. Brief. Bioinform. 2021, 22, bbab174. [Google Scholar] [CrossRef]
- Liao, Q.; Ye, Y.; Li, Z.; Chen, H.; Zhuo, L. Prediction of miRNA-disease associations in microbes based on graph convolutional networks and autoencoders. Front. Microbiol. 2023, 14, 1170559. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Ding, P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 2018, 34, 239–248. [Google Scholar] [CrossRef]
- Lee, I.; Blom, U.M.; Wang, P.I.; Shim, J.; Marcotte, E.M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011, 21, 1109–1121. [Google Scholar] [CrossRef]
- Dai, Q.; Wang, Z.; Liu, Z.; Duan, X.; Song, J.; Guo, M. Predicting miRNA-disease associations using an ensemble learning framework with resampling method. Brief. Bioinform. 2022, 23, bbab543. [Google Scholar] [CrossRef]
- Li, Z.; Zhong, T.; Huang, D.; You, Z.-H.; Nie, R. Hierarchical graph attention network for miRNA-disease association prediction. Mol. Ther. 2022, 30, 1775–1786. [Google Scholar] [CrossRef]
- Zhang, H.; Fang, J.; Sun, Y.; Xie, G.; Lin, Z.; Gu, G. Predicting miRNA-Disease Associations via Node-Level Attention Graph Auto-Encoder. Ieee-Acm Trans. Comput. Biol. Bioinform. 2023, 20, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Cheng, Z.; Li, H.; Teng, Z.; Liu, Y.; Tian, Z. Predicting miRNA-disease associations via learning multimodal networks and fusing mixed neighborhood information. Brief. Bioinform. 2022, 23, bbac159. [Google Scholar] [CrossRef]
- Wang, W.; Chen, H. Predicting miRNA-disease associations based on lncRNA-miRNA interactions and graph convolution networks. Brief. Bioinform. 2022, 24, bbac495. [Google Scholar] [CrossRef]
- Ning, Q.; Zhao, Y.; Gao, J.; Chen, C.; Li, X.; Li, T.; Yin, M. AMHMDA: Attention aware multi-view similarity networks and hypergraph learning for miRNA-disease associations identification. Brief. Bioinform. 2023, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.-M.; Lin, J.-T.; Huang, S.-P.; Chiu, H.-M.; Wang, H.-P.; Lee, Y.-C.; Lin, J.-W.; Shun, C.; Liang, J.-T.; Wu, M.-S. Screening for colorectal cancer in average-risk Chinese population using a mixed strategy with sigmoidoscopy and colonoscopy. Dis. Colon Rectum 2007, 50, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Uddin, A.; Chakraborty, S. Role of miRNAs in lung cancer. J. Cell. Physiol. 2018, 1–10. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. dbDEMC 2.0: Updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Z.; Zhu, R.; Liu, J.; Shang, J.; Dai, L. HGSMDA: miRNA–Disease Association Prediction Based on HyperGCN and Sørensen-Dice Loss. Non-Coding RNA 2024, 10, 9. https://doi.org/10.3390/ncrna10010009
Chang Z, Zhu R, Liu J, Shang J, Dai L. HGSMDA: miRNA–Disease Association Prediction Based on HyperGCN and Sørensen-Dice Loss. Non-Coding RNA. 2024; 10(1):9. https://doi.org/10.3390/ncrna10010009
Chicago/Turabian StyleChang, Zhenghua, Rong Zhu, Jinxing Liu, Junliang Shang, and Lingyun Dai. 2024. "HGSMDA: miRNA–Disease Association Prediction Based on HyperGCN and Sørensen-Dice Loss" Non-Coding RNA 10, no. 1: 9. https://doi.org/10.3390/ncrna10010009
APA StyleChang, Z., Zhu, R., Liu, J., Shang, J., & Dai, L. (2024). HGSMDA: miRNA–Disease Association Prediction Based on HyperGCN and Sørensen-Dice Loss. Non-Coding RNA, 10(1), 9. https://doi.org/10.3390/ncrna10010009