

lncRNA-mRNA Co-Expression and Regulation Analysis in Lung Fibroblasts from Idiopathic Pulmonary Fibrosis


 , and
, and
Abstract
:1. Introduction
2. Results
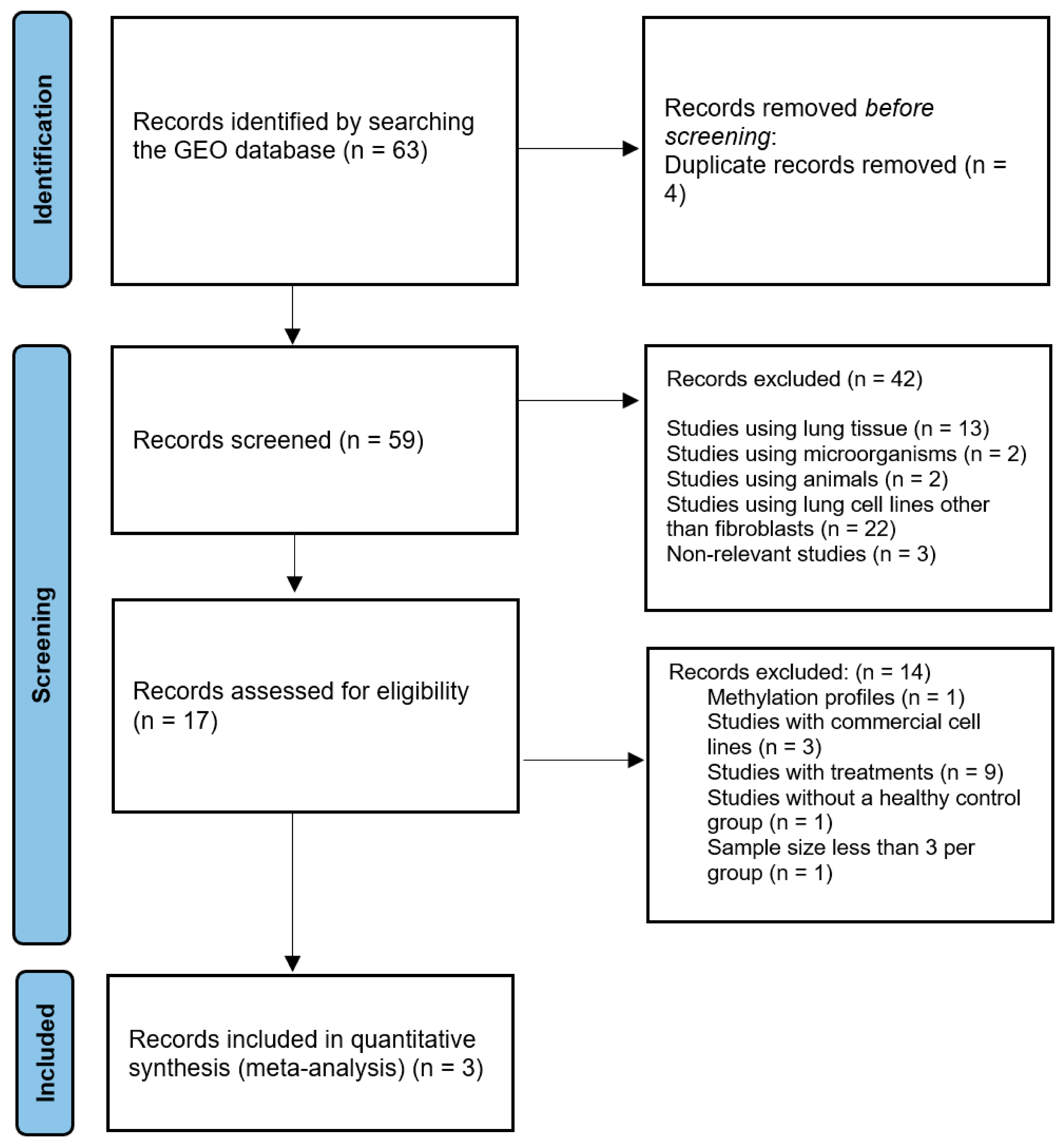
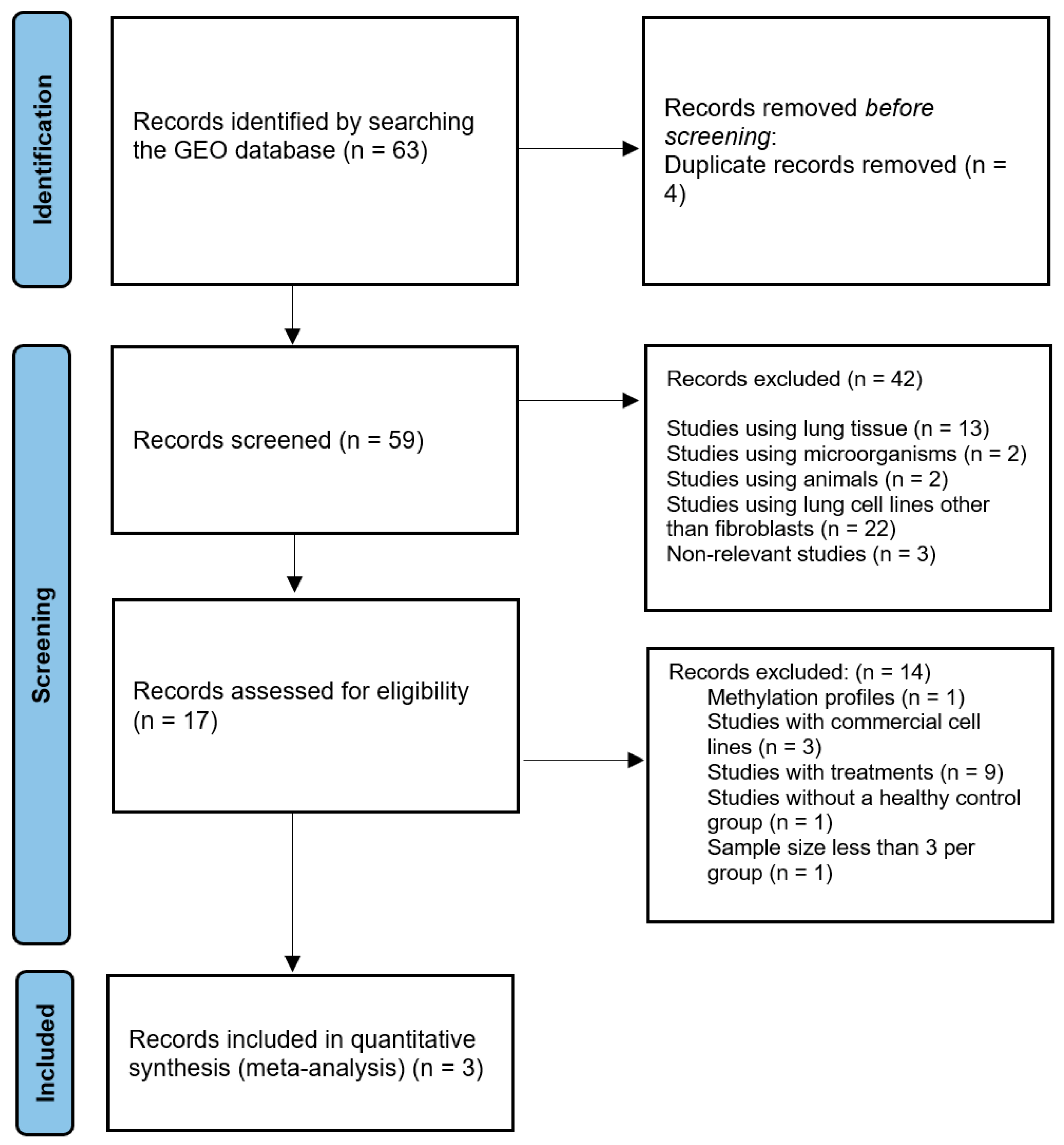
2.1. Study Selection
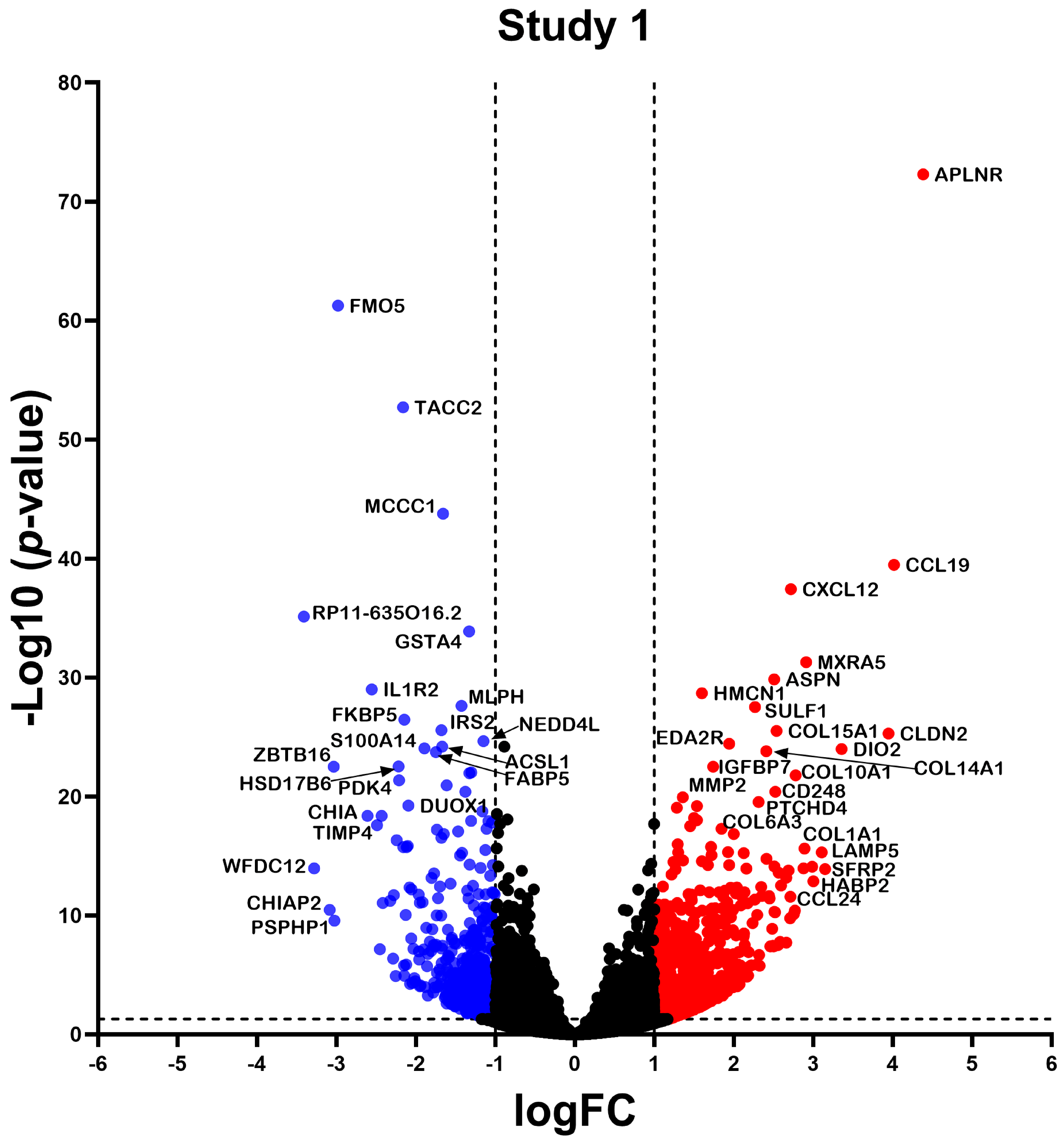
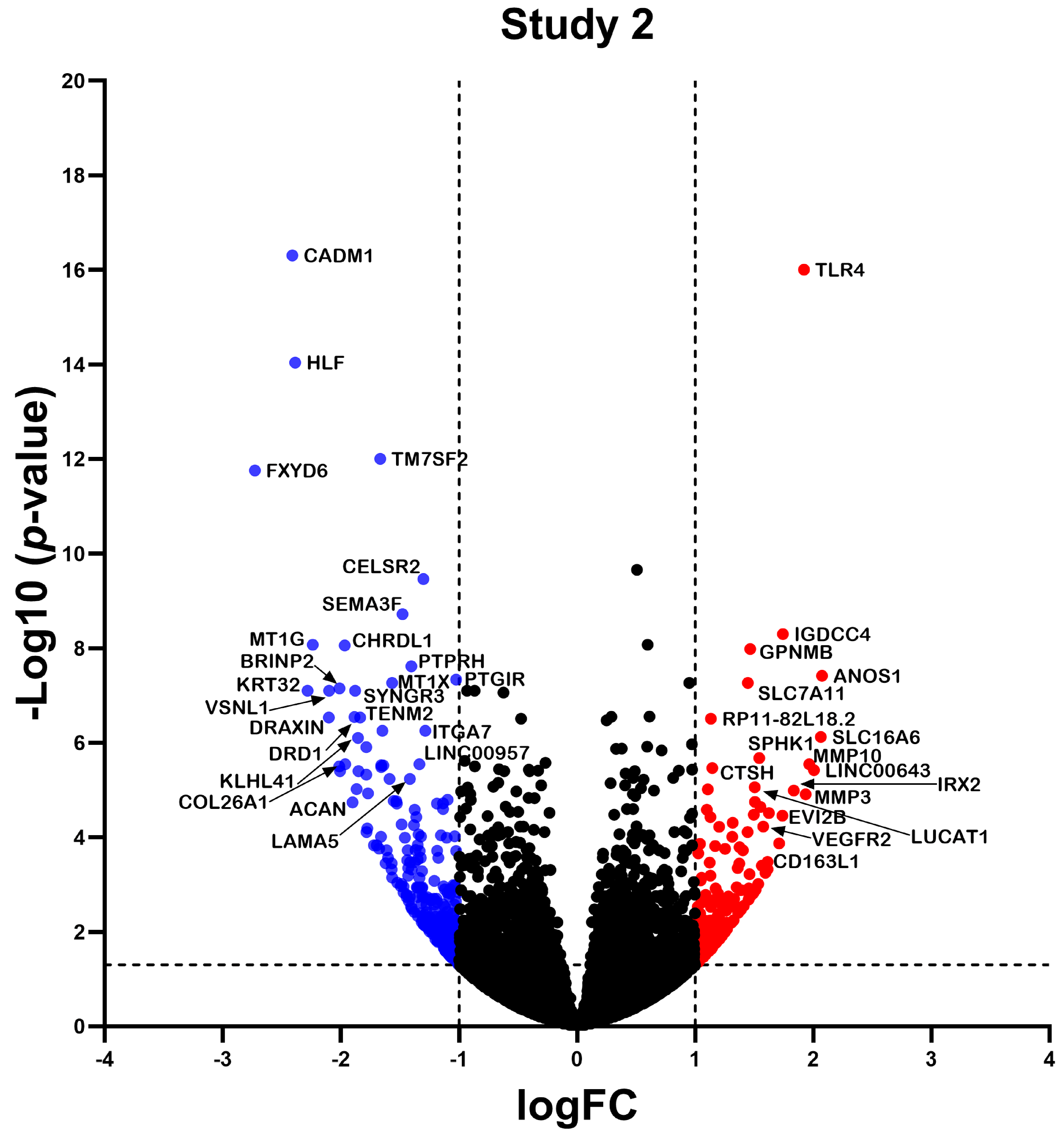
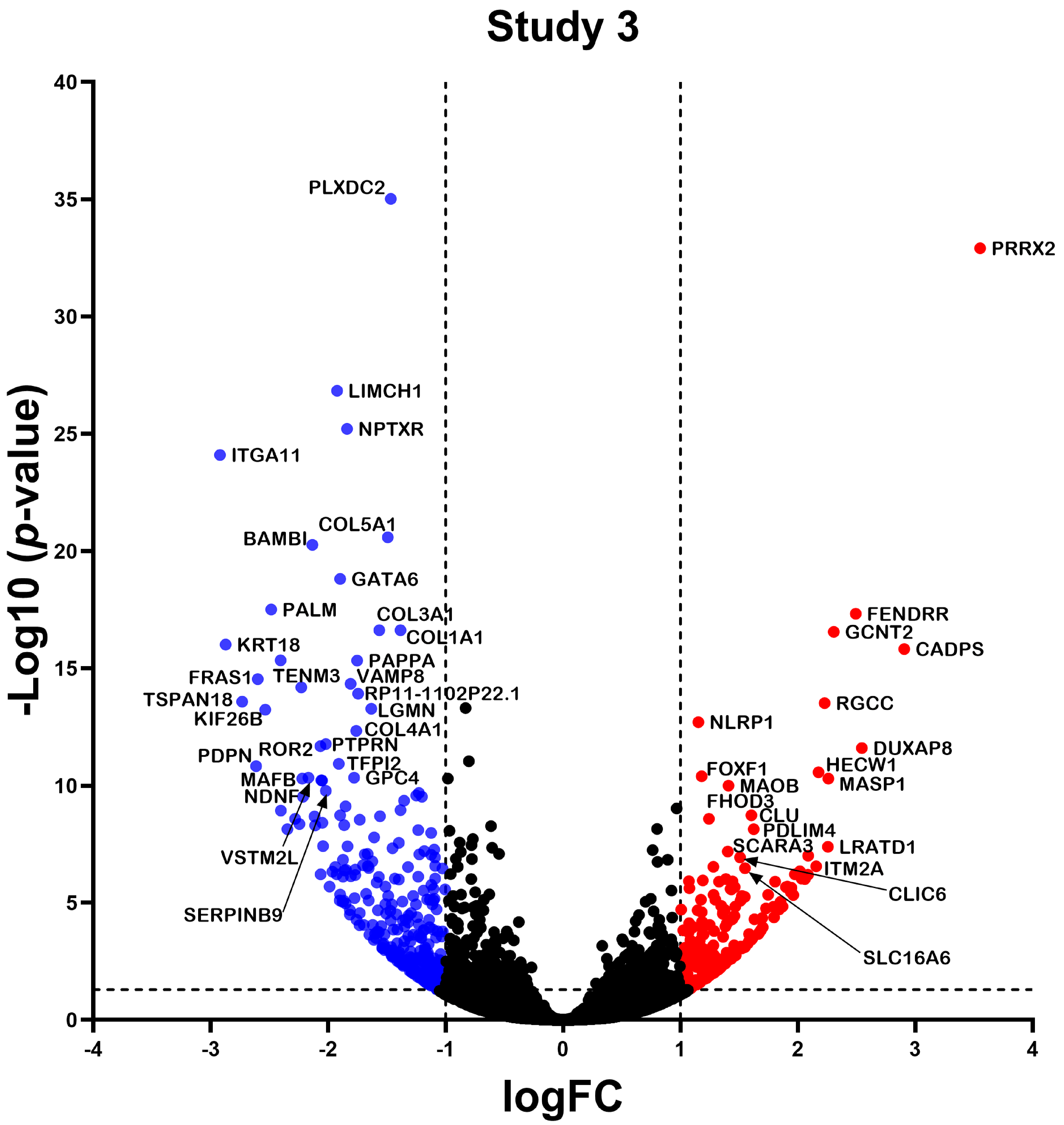
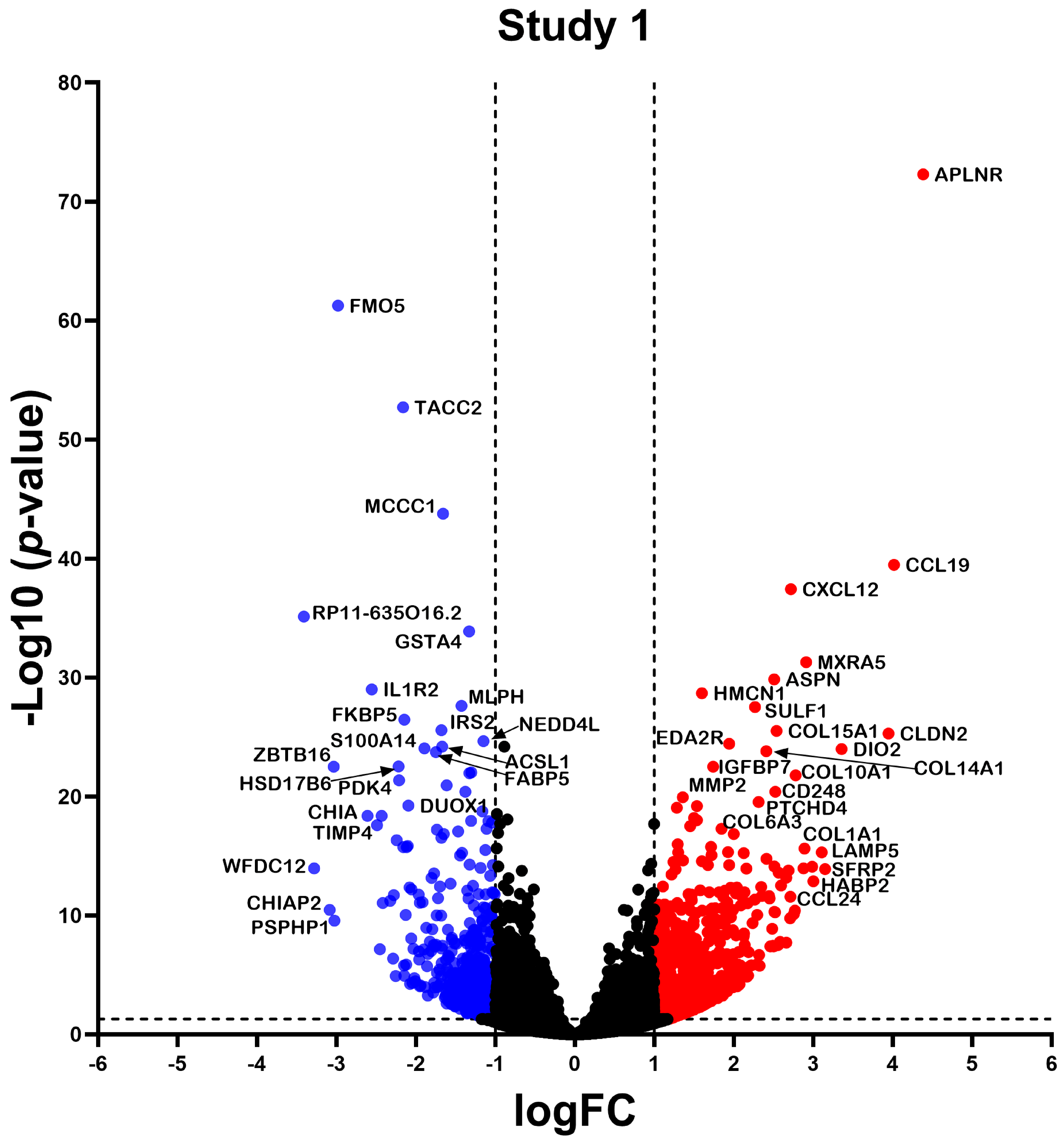
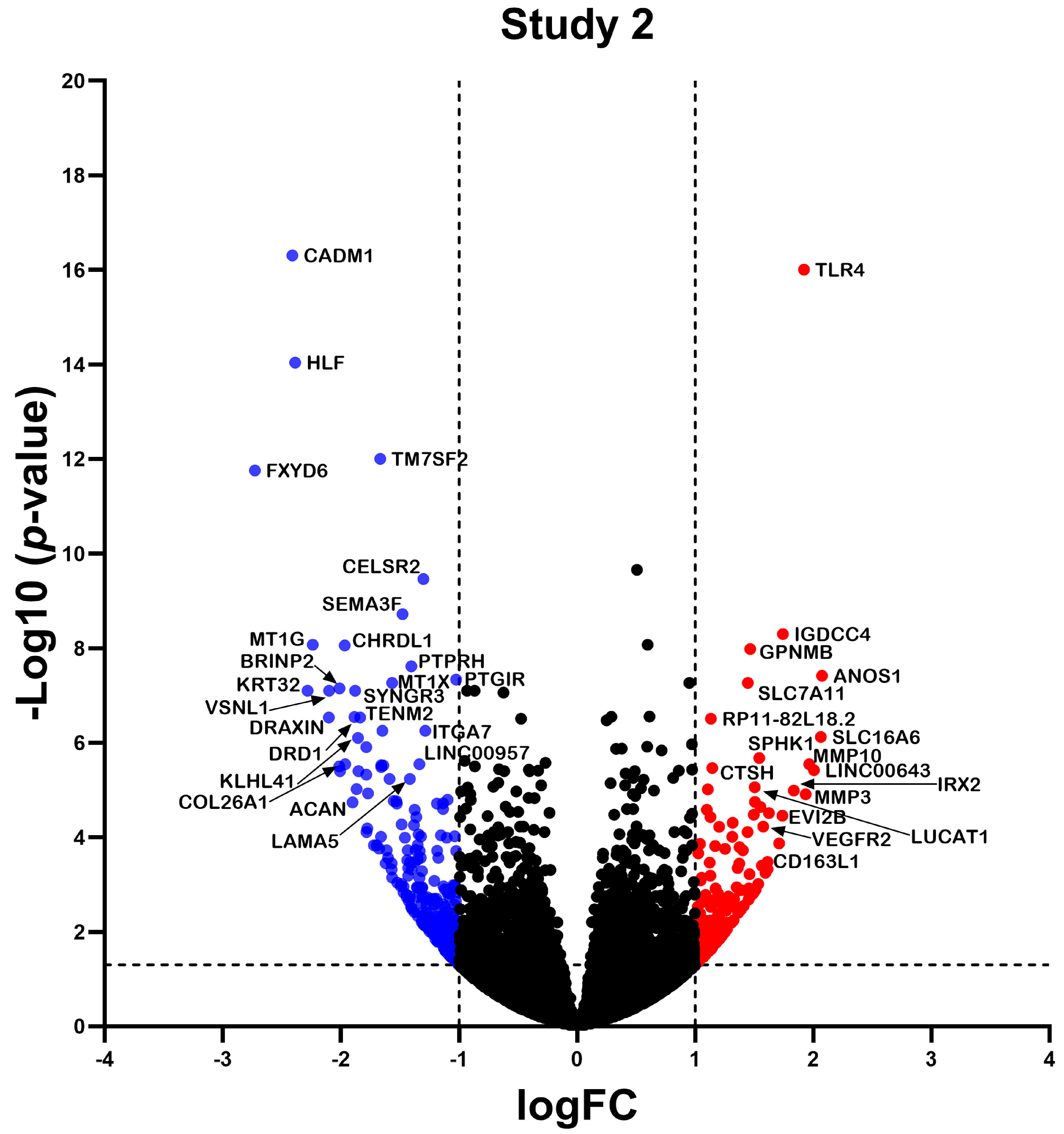
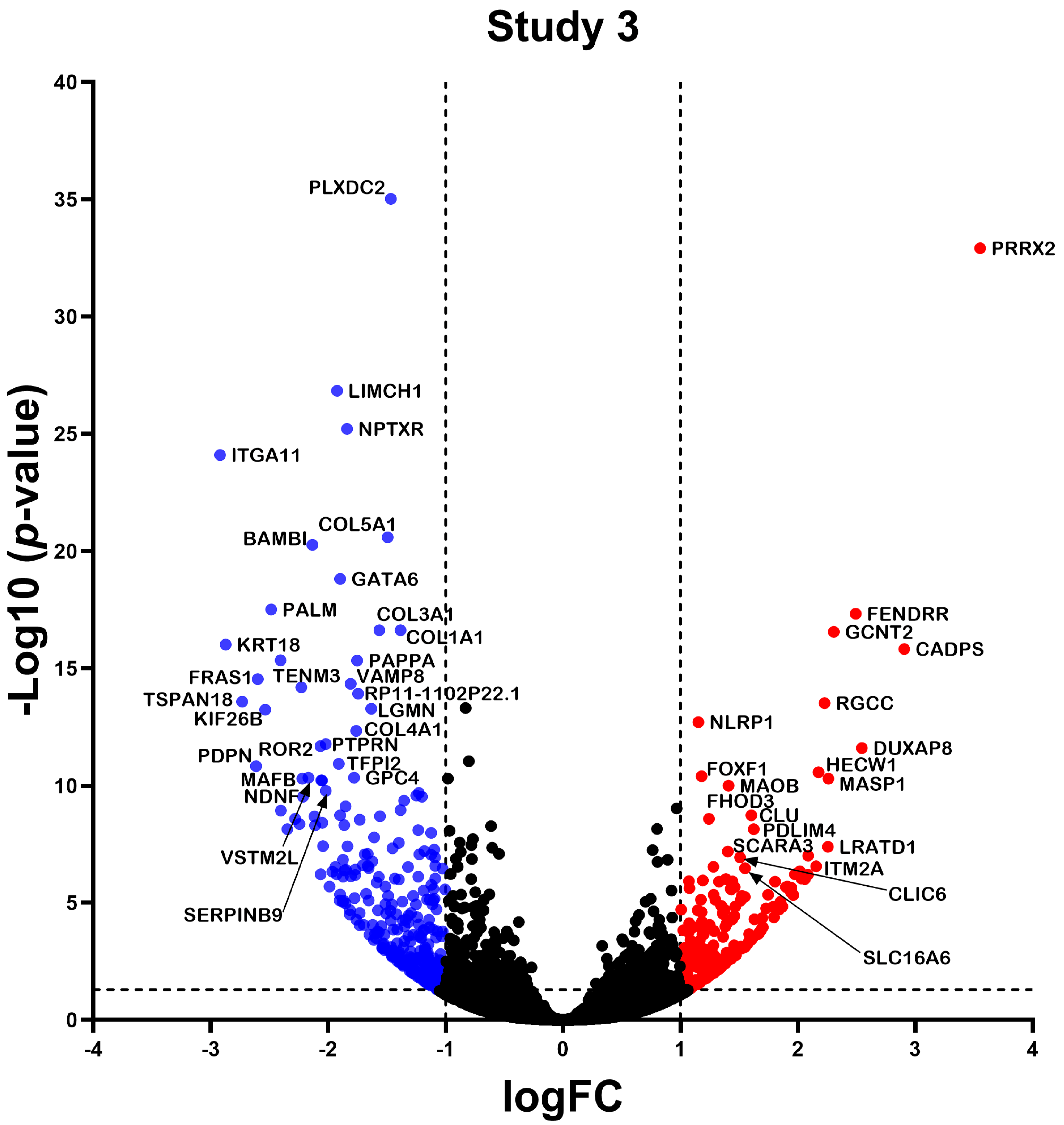
2.2. Differentially Expressed Gene
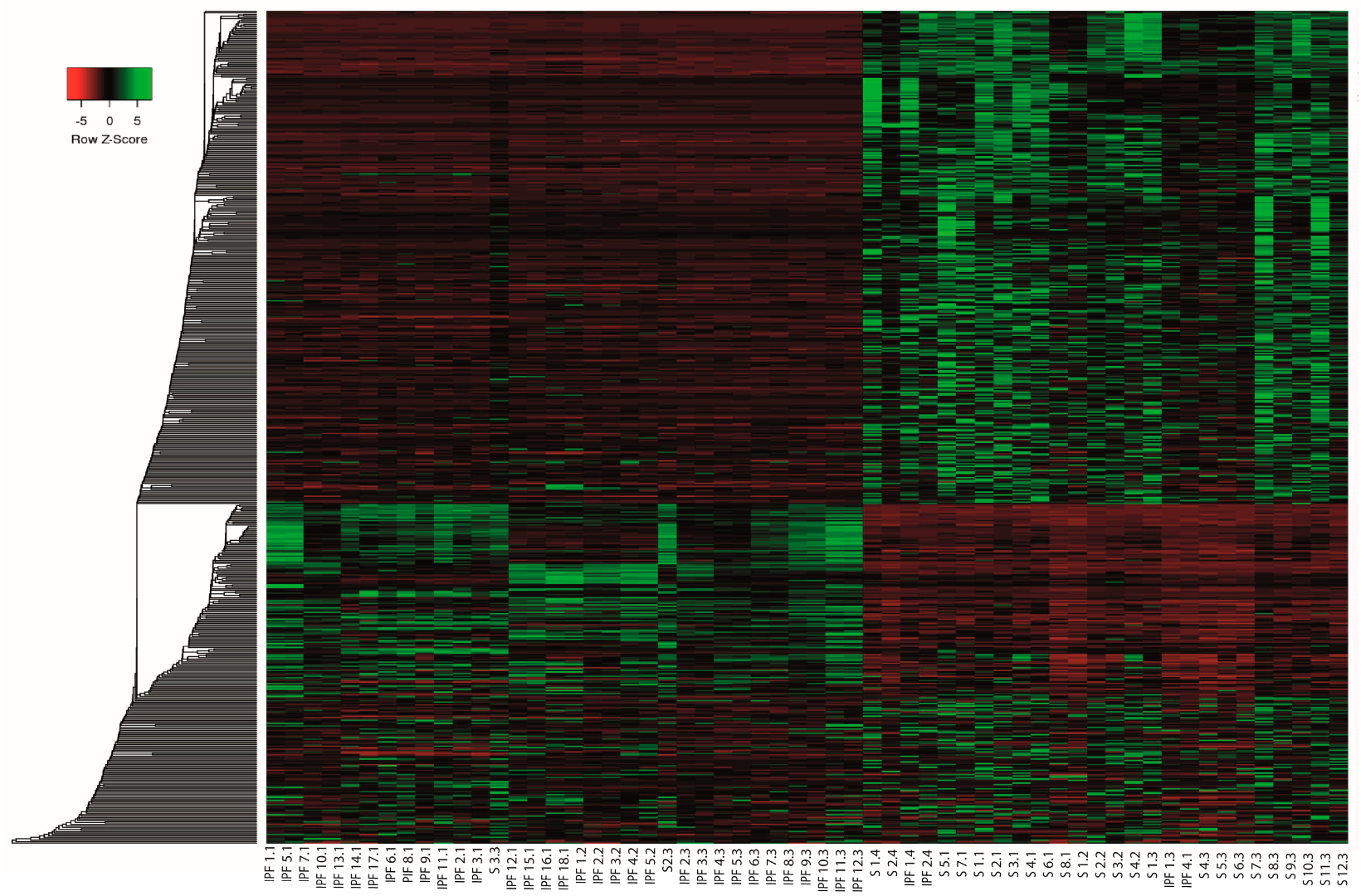
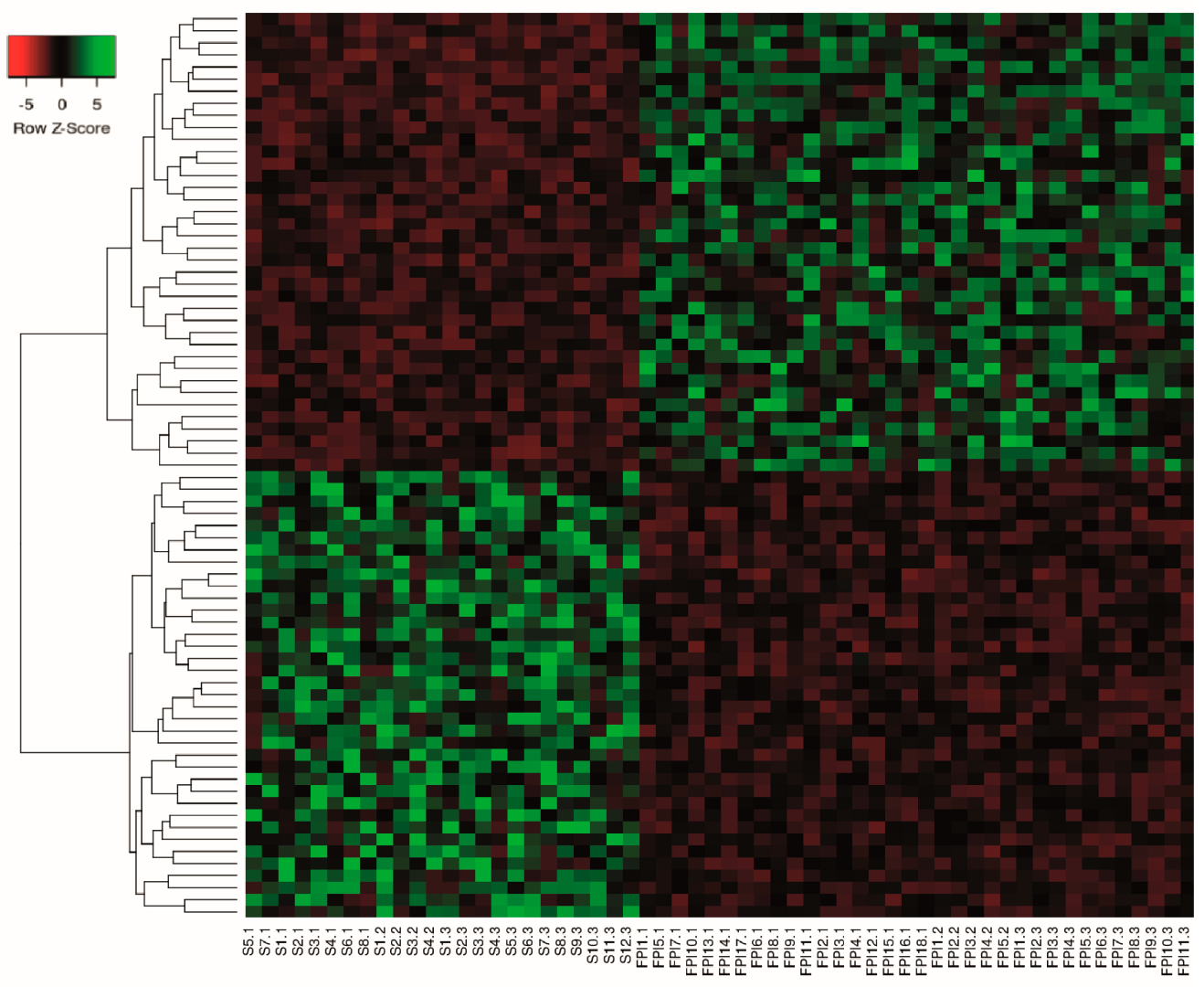
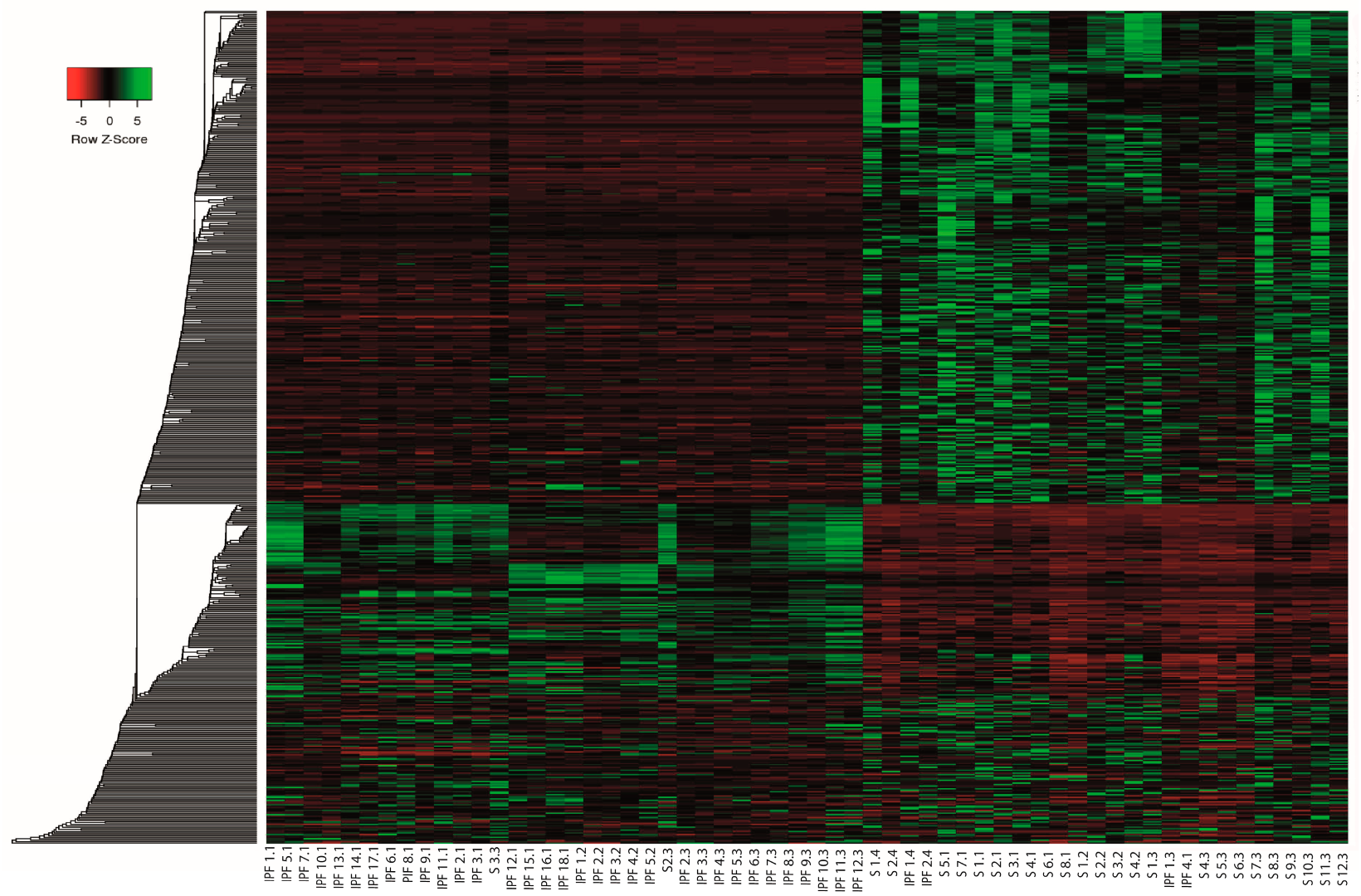
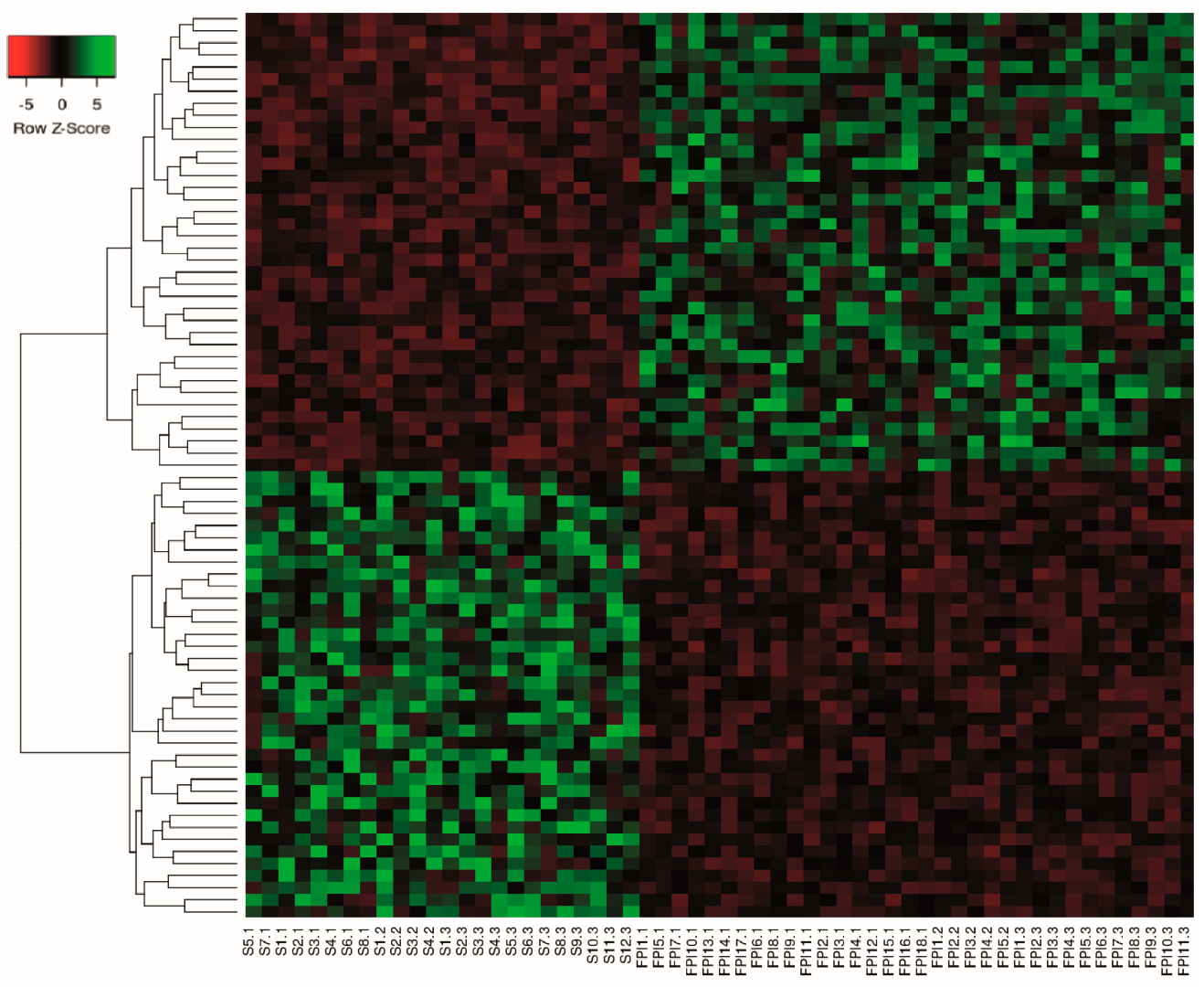
2.3. Differential Expression Analysis of LncRNAs and mRNAs
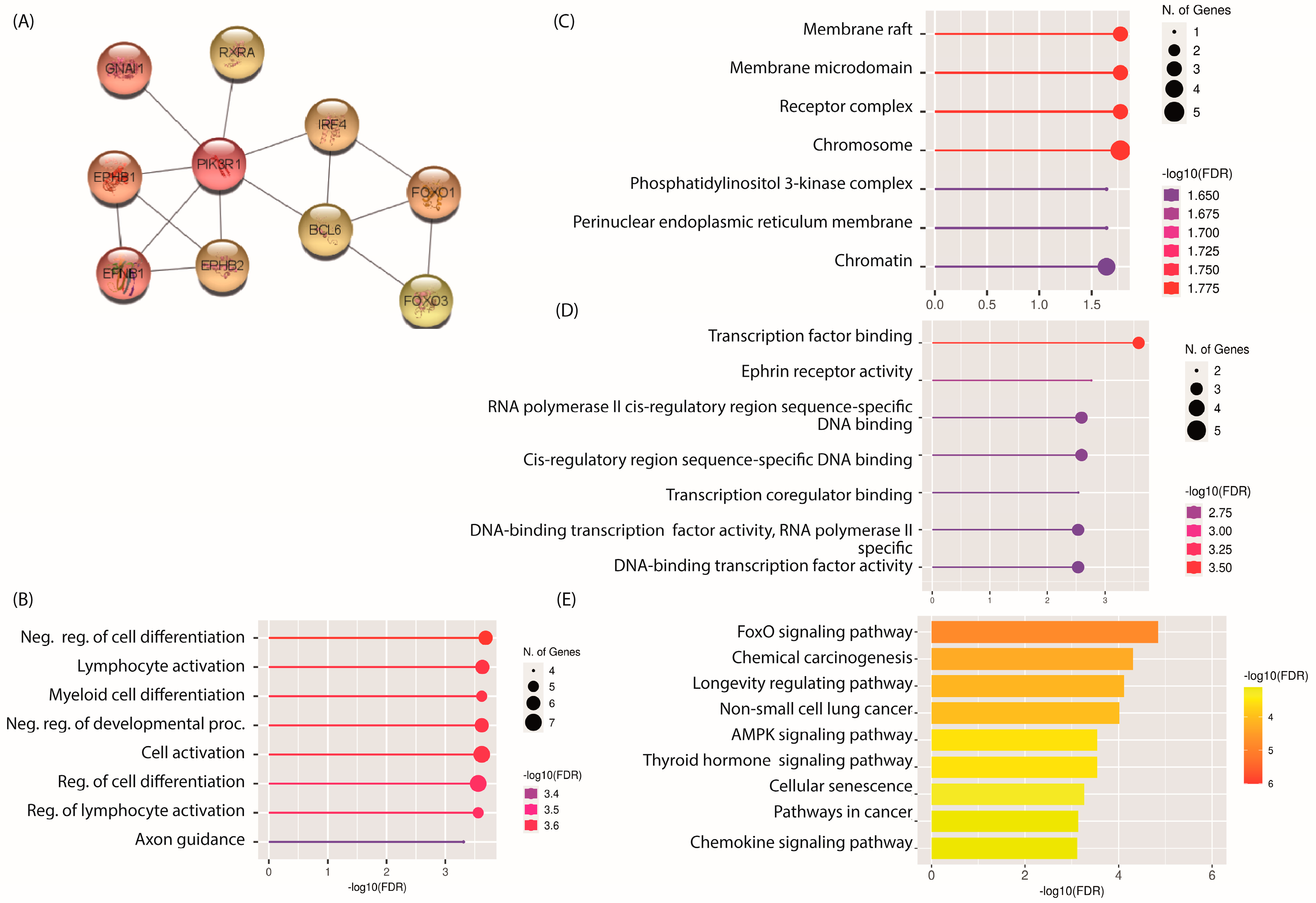
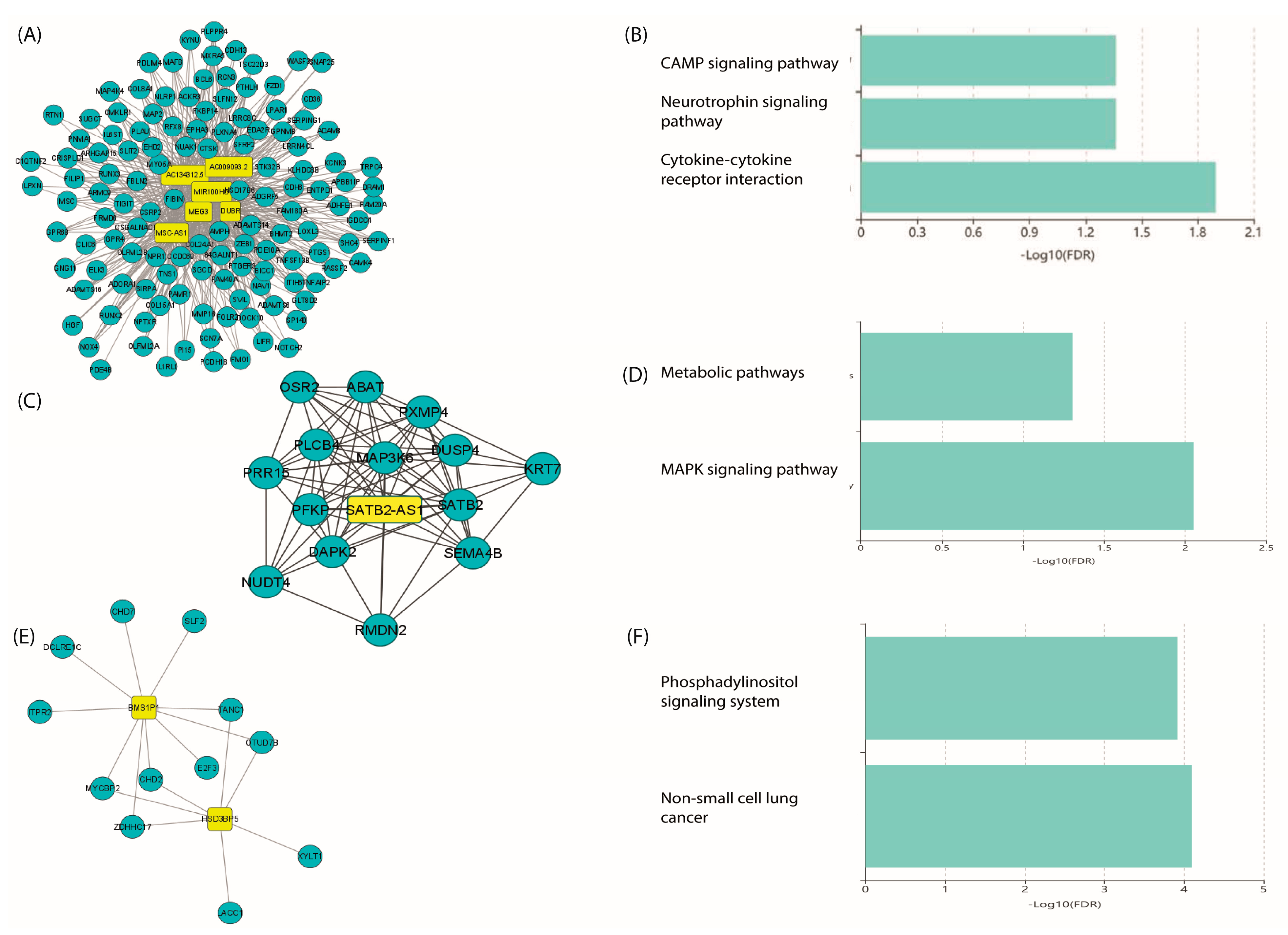
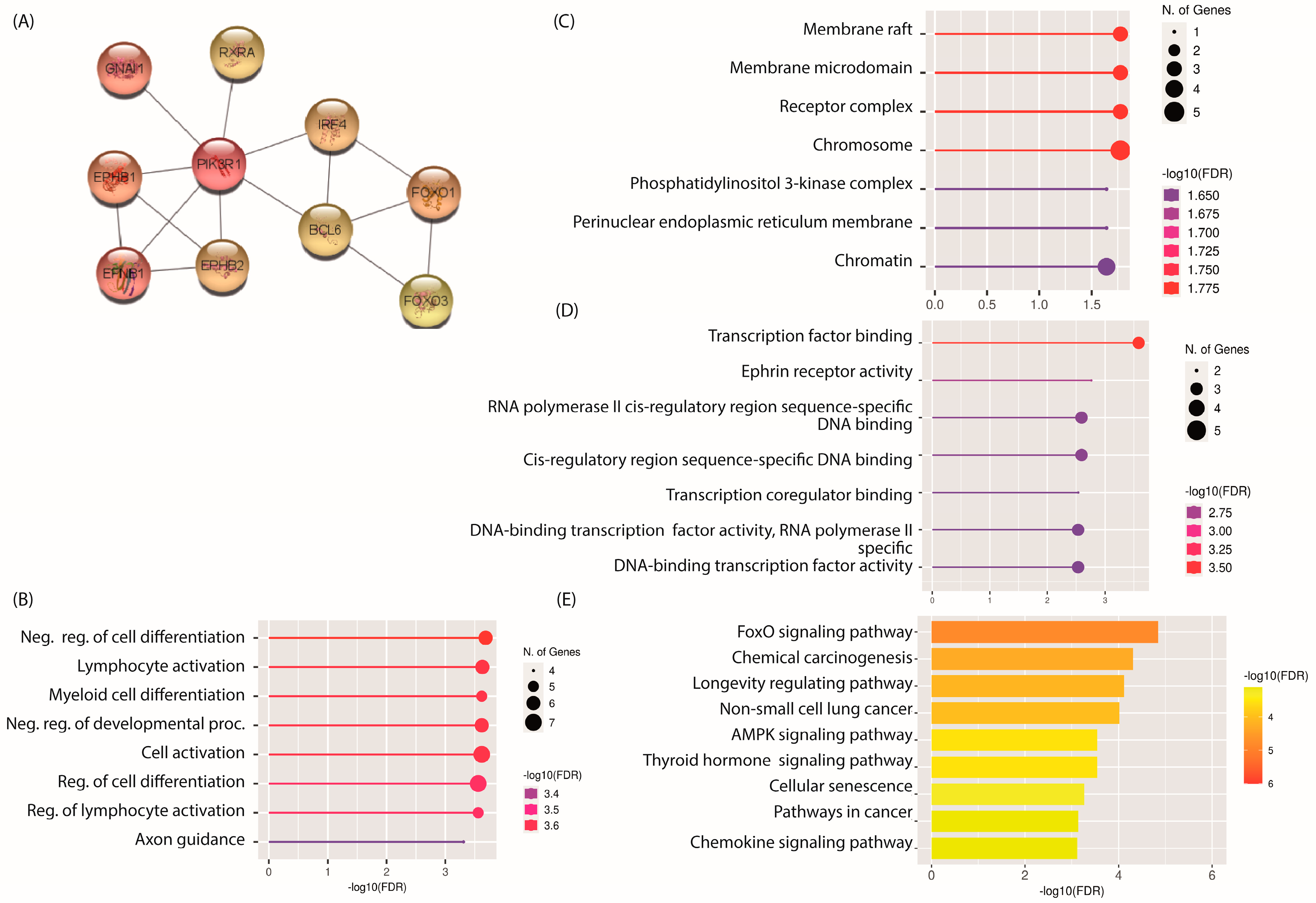
2.4. PPI Network and Identification of Hub mRNAs
2.5. GO and KEGG Pathway Analysis of Hub mRNAs
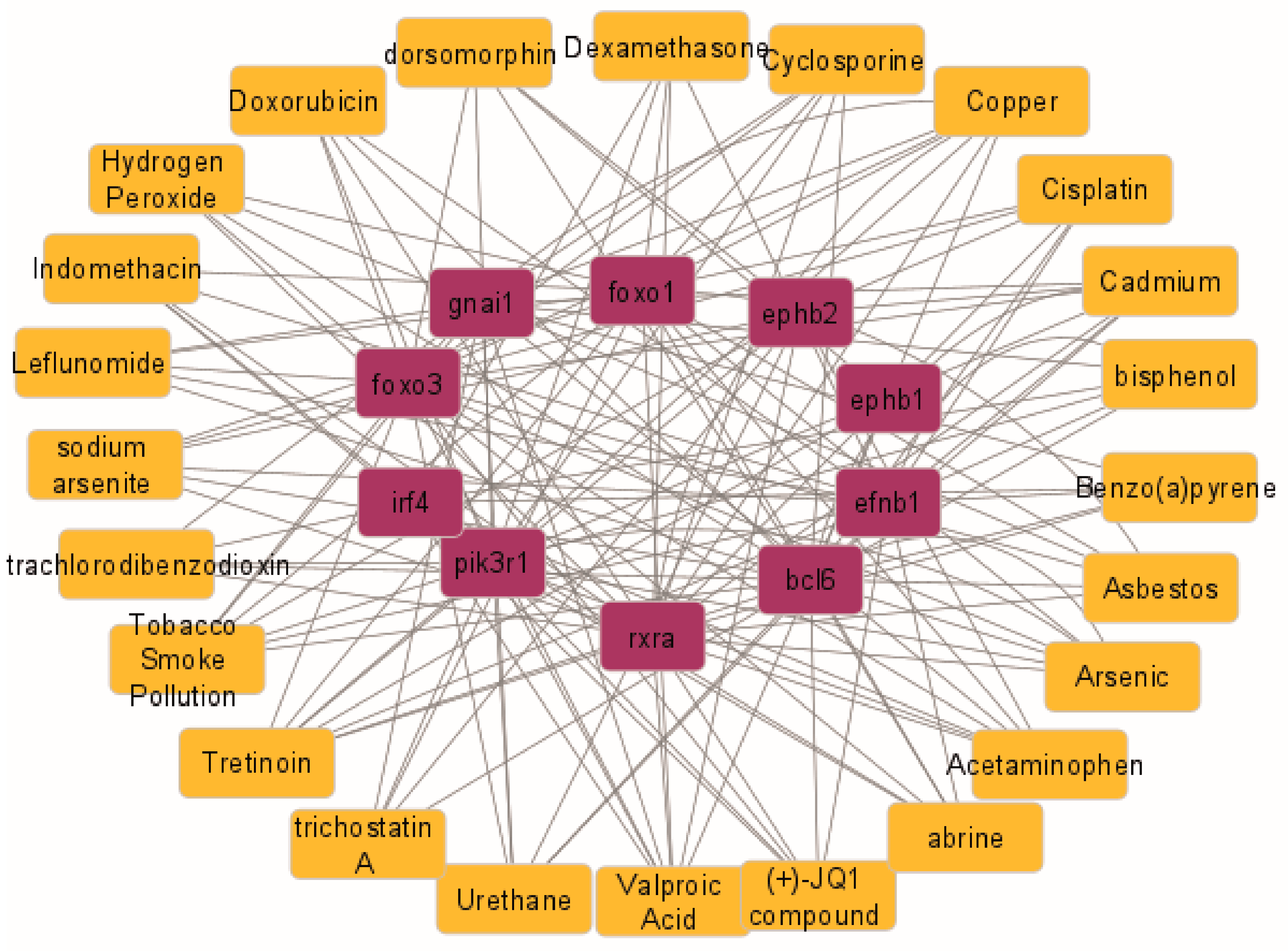
2.6. Hub Gene–Drug Interaction Network Analysis
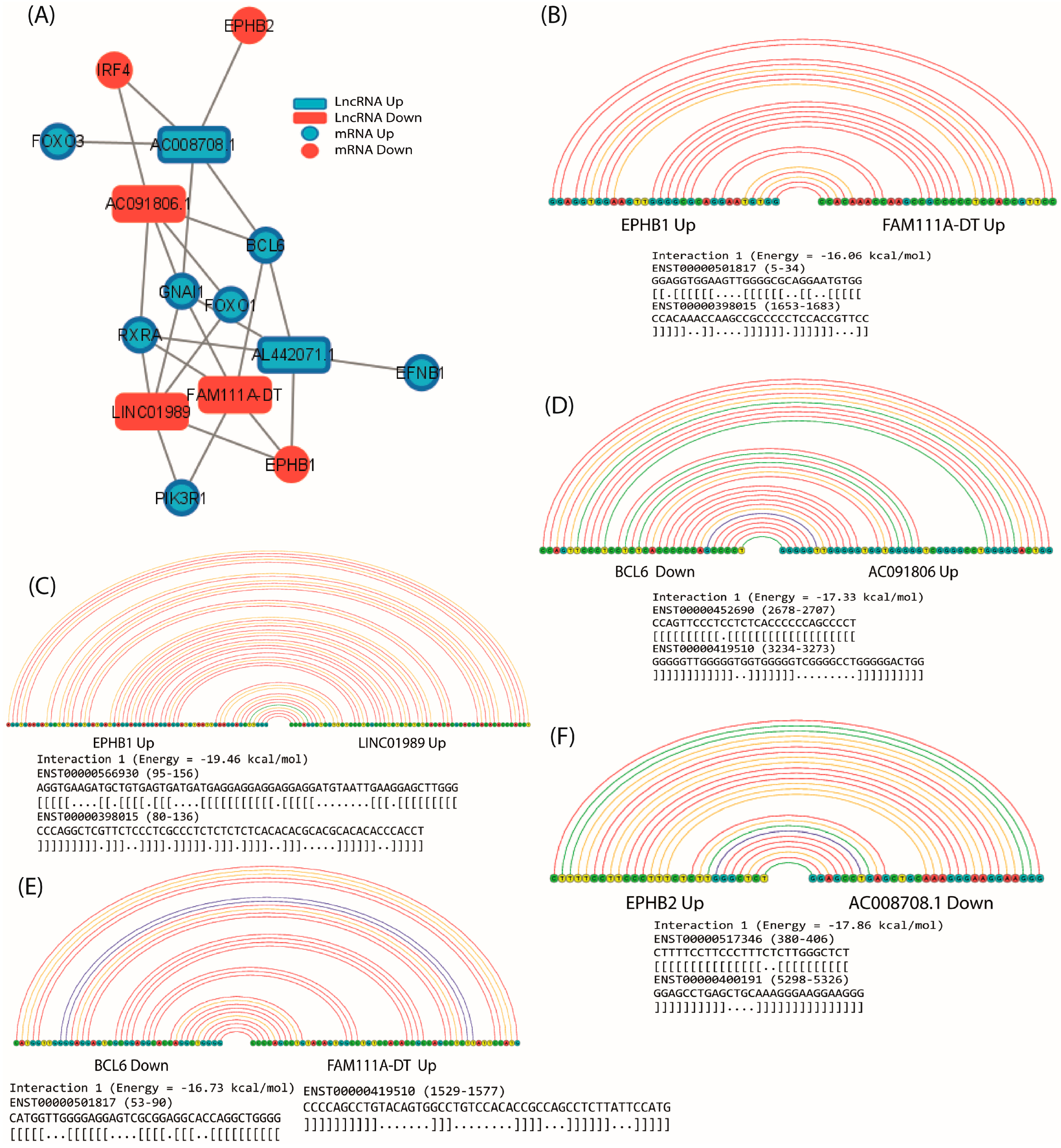
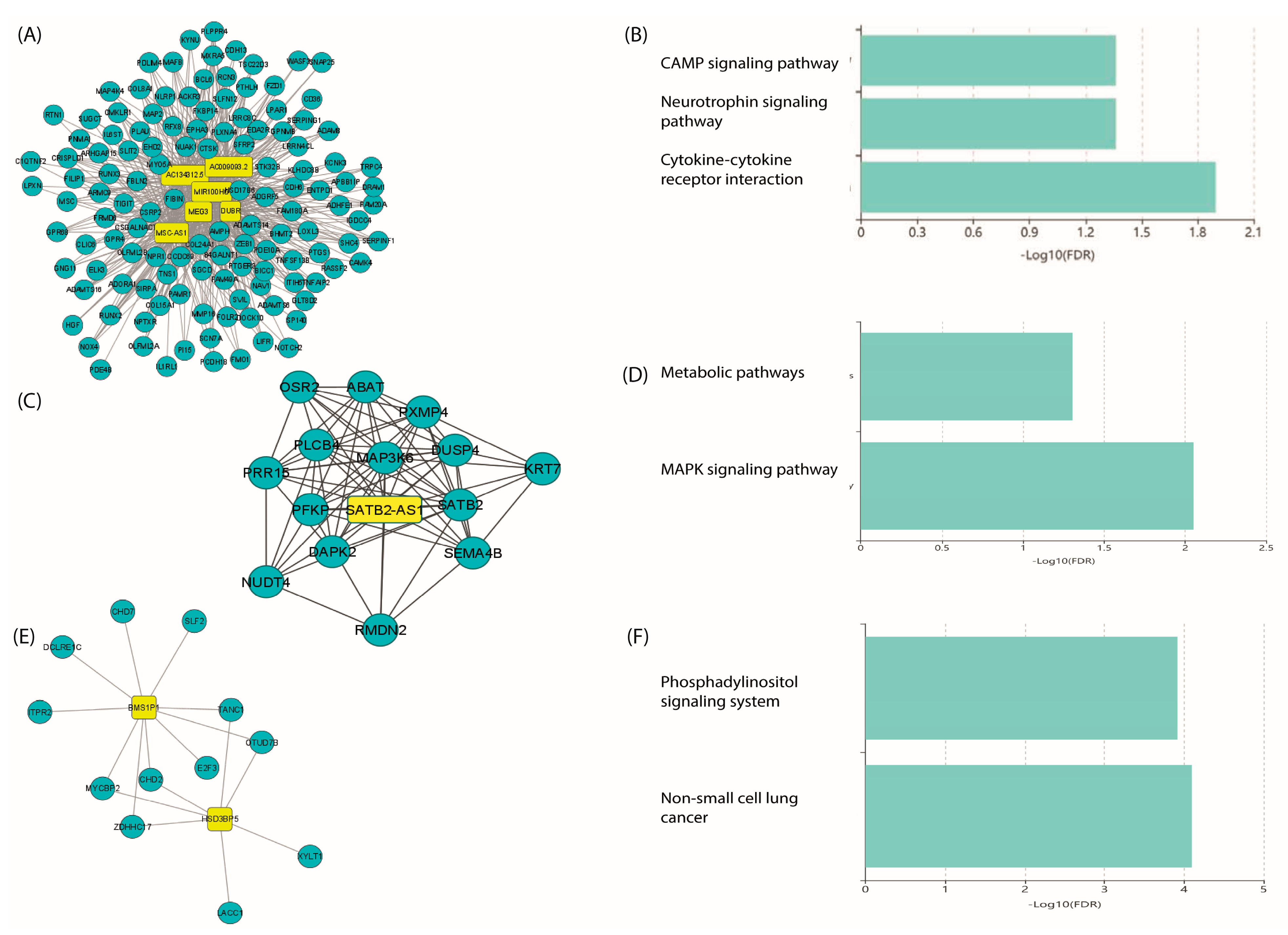
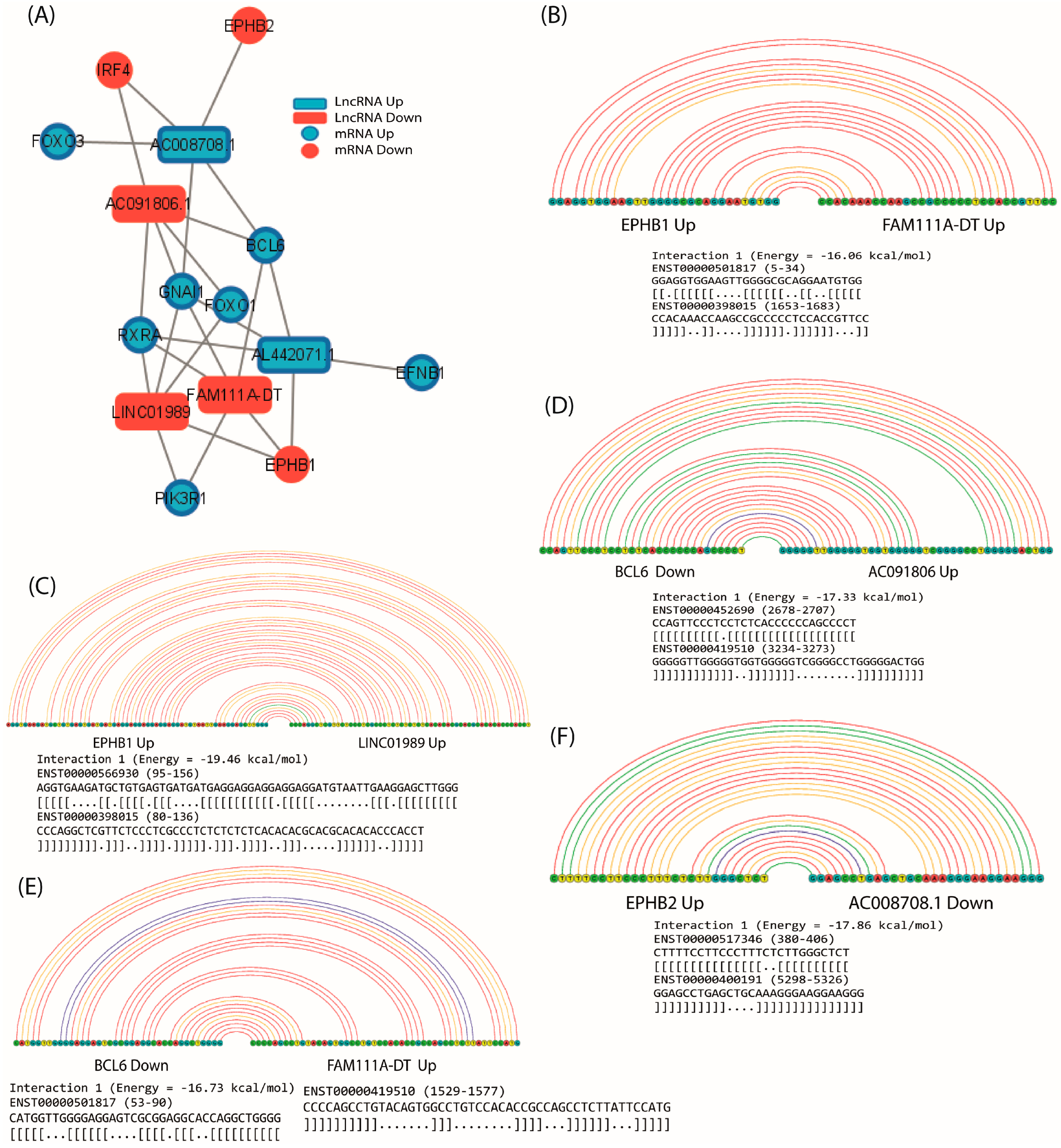
2.7. Co-Expression of LncRNA/mRNAs
2.8. Prediction of lncRNA–mRNA Interactions
3. Discussion
4. Materials and Methods
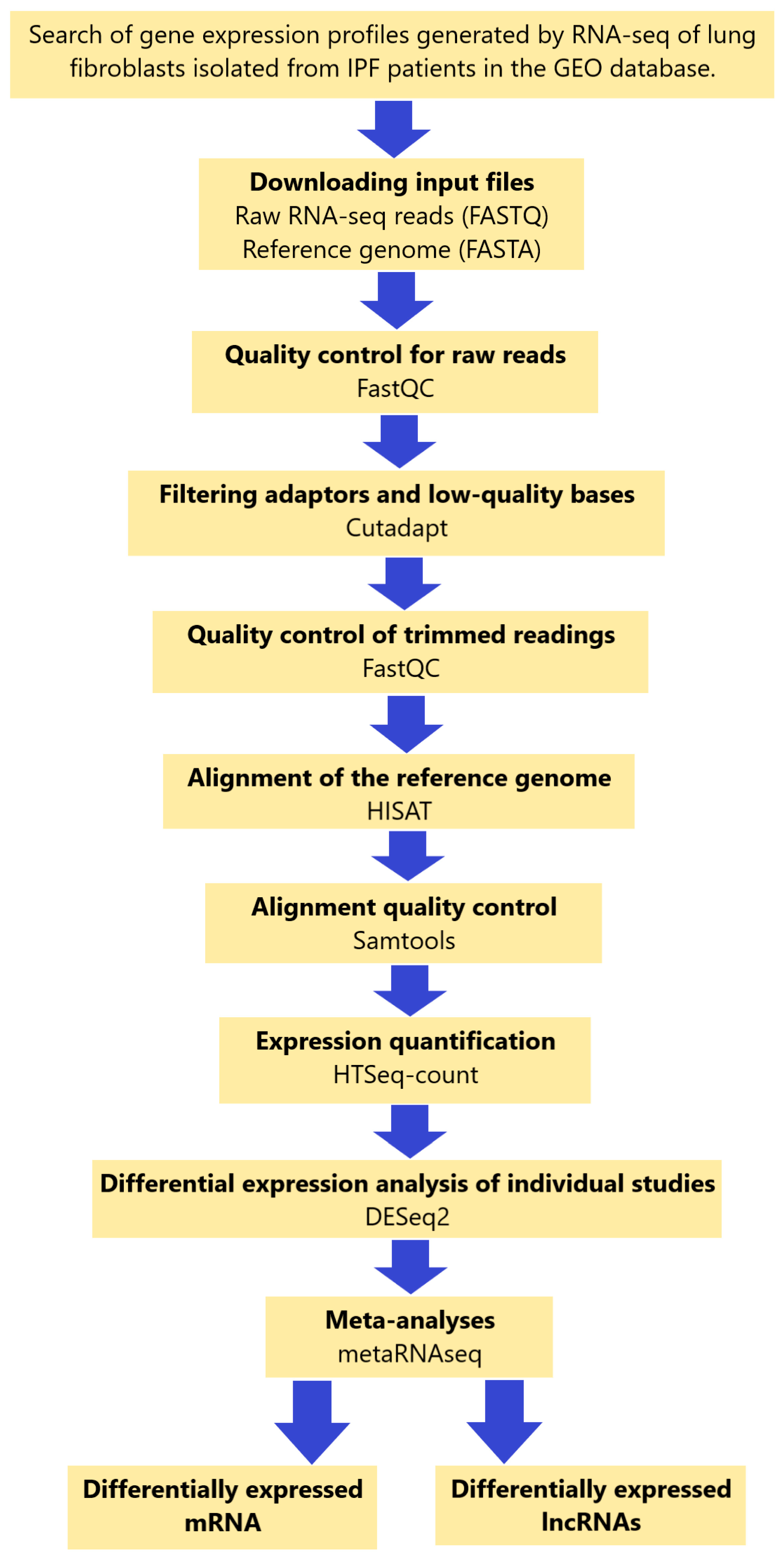
4.1. Search and Selection Criteria for RNA-Seq Datasets
- Studies in primary fibroblasts isolated from human lung tissue.
- Expression profiles generated by high-throughput sequencing (RNA-seq).
- Comparative study between primary fibroblasts isolated from lung tissues of patients with IPF, as well as their respective controls; the latter consisting of primary fibroblasts isolated from normal human lung tissues.
- Sample size equal to or more than 3 per group.
- The raw RNA-seq-generated reads are publicly available.
- Expression profiles generated by microarrays are not included.
- Expression profiles from commercial cell lines are excluded.
- Lung tissue samples are excluded.
- Expression profiles from primary cultured fibroblasts isolated from species other than Homo sapiens are not considered.
- RNA-seq profiles are not accepted if raw data are not publicly available.
- Methylation profiles are excluded.
- The meta-analysis was conducted following the guidelines provided in the 2020 Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [72].
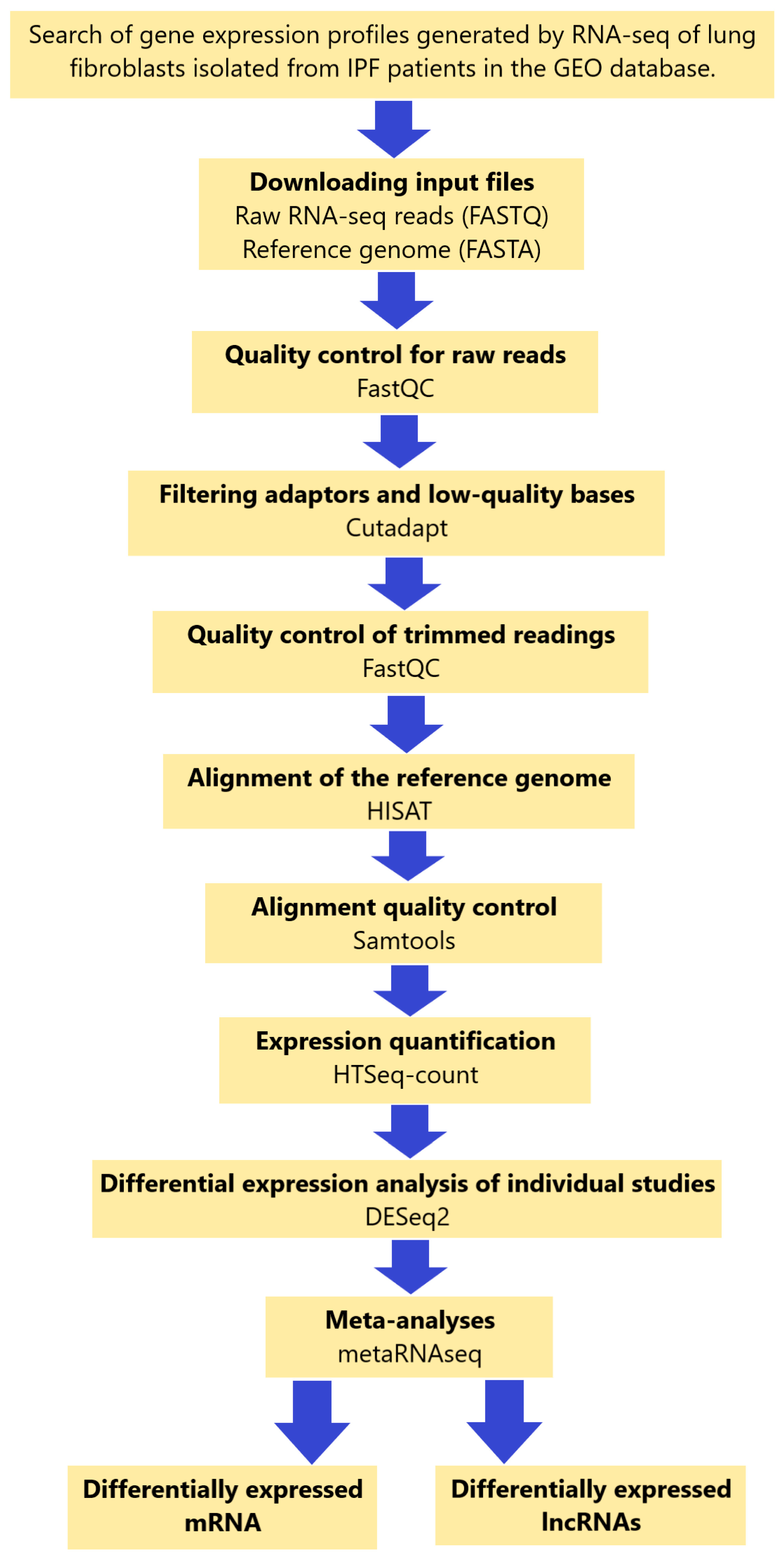
4.2. Data Collection
4.3. Data Analysis of Individual Study Data
4.4. Alignment of Transcripts with the Reference Genome
4.5. Differentially Expressed Analysis
4.6. Meta-Analysis
4.7. Gene Ontology and KEGG Pathway Enrichment Analysis
4.8. Protein–Protein Interaction Network
4.9. Comparative Toxicogenomic Interaction Network
4.10. lncRNA–mRNA Network Construction
4.11. Prediction of lncRNA–mRNA Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wakwaya, Y.; Brown, K.K. Idiopathic Pulmonary Fibrosis: Epidemiology, Diagnosis and Outcomes. Am. J. Med. Sci. 2019, 357, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Hu, Y. TGF-β1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. [Google Scholar] [CrossRef] [PubMed]
- Vivien, S.; Nazia, C.; Sebastiano Emanuele, T.; Nicolas, K.; Veronika, M.; Michael, K. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 190021. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Li, C.; Zhang, J.; Su, Z. Chemotherapy in idiopathic pulmonary fibrosis and small-cell lung cancer with poor lung function. BMC Pulm. Med. 2021, 21, 122. [Google Scholar] [CrossRef] [PubMed]
- Lanzafame, M.; Bianco, G.; Terracciano, L.M.; Ng, C.K.Y.; Piscuoglio, S. The Role of Long Non-Coding RNAs in Hepatocarcinogenesis. Int. J. Mol. Sci. 2018, 19, 682. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, M.; Kasprowicz, M.K.; Kasprzyk, M.E.; Wrzesinski, J. Human Long Noncoding RNA Interactome: Detection, Characterization and Function. Int. J. Mol. Sci. 2020, 21, 1027. [Google Scholar] [CrossRef] [PubMed]
- Hadjicharalambous, M.R.; Roux, B.T.; Csomor, E.; Feghali-Bostwick, C.A.; Murray, L.A.; Clarke, D.L.; Lindsay, M.A. Long intergenic non-coding RNAs regulate human lung fibroblast function: Implications for idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 6020. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jiao, B.; Gui, W.; Zhang, Q.; Wang, F.; Han, L. Long non-coding RNA SNHG1 promotes fibroblast-to-myofibroblast transition during the development of pulmonary fibrosis induced by silica particles exposure. Ecotoxicol. Environ. Saf. 2021, 228, 112938. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Tiret, L. Gene-environment interaction: A central concept in multifactorial diseases. Proc. Nutr. Soc. 2002, 61, 457–463. [Google Scholar] [CrossRef]
- Hamadeh, H.K.; Amin, R.P.; Paules, R.S.; Afshari, C.A. An overview of toxicogenomics. Curr. Issues Mol. Biol. 2002, 4, 45–56. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.Z.; Guan, N.N.; Qu, J.; Huang, Z.A.; Zhu, Z.X.; Li, J.Q. Computational models for lncRNA function prediction and functional similarity calculation. Brief. Funct. Genom. 2019, 18, 58–82. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Satagopam, V.; Schneider, R. Long Non-coding RNAs: Mechanisms, Experimental, and Computational Approaches in Identification, Characterization, and Their Biomarker Potential in Cancer. Front. Genet. 2021, 12, 649619. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mendoza, L.; Monzon-Sandoval, J.; Urrutia, A.O.; Gutierrez, H. Emergence of co-expression in gene regulatory networks. PLoS ONE 2021, 16, e0247671. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Hamada, M. RIblast: An ultrafast RNA-RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Han, H.; Lagares, D. RNA Sequencing Uncovers Antifibrotic Genes during Lung Fibrosis Resolution. Am. J. Respir. Cell Mol. Biol. 2021, 64, 401–402. [Google Scholar] [CrossRef]
- Pham, T.X.; Lee, J.; Guan, J.; Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Huang, S.K.; Tschumperlin, D.J.; Ligresti, G. Transcriptional analysis of lung fibroblasts identifies PIM1 signaling as a driver of aging-associated persistent fibrosis. JCI Insight 2022, 7, e153672. [Google Scholar] [CrossRef]
- Mullenbrock, S.; Liu, F.; Szak, S.; Hronowski, X.; Gao, B.; Juhasz, P.; Sun, C.; Liu, M.; McLaughlin, H.; Xiao, Q.; et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes 2018, 9, 588. [Google Scholar] [CrossRef]
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of Cultured Lung Fibroblasts in Idiopathic Pulmonary Fibrosis: Meta-Analysis of Publically Available Microarray Datasets Reveals Repression of Inflammation and Immunity Pathways. Int. J. Mol. Sci. 2016, 17, 2091. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Xue, H.; Wei, L.; Jin, J.; Tian, X.; Wang, Q. High heterogeneity undermines generalization of differential expression results in RNA-Seq analysis. Hum. Genom. 2021, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Snyder, M.P. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e219. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, Q.-q.; Liao, H.-h.; Fan, D.; Tang, Q.-z. Bcl6 Suppresses Cardiac Fibroblast Activation and Function via Directly Binding to Smad4. Curr. Med. Sci. 2019, 39, 534–540. [Google Scholar] [CrossRef]
- Salbert, G.; Fanjul, A.; Piedrafita, F.J.; Lu, X.P.; Kim, S.J.; Tran, P.; Pfahl, M. Retinoic acid receptors and retinoid X receptor-alpha down-regulate the transforming growth factor-beta 1 promoter by antagonizing AP-1 activity. Mol. Endocrinol. (Baltim. Md.) 1993, 7, 1347–1356. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Zheng, Y.; Chen, W.; Sun, Y.; Wu, Z.; Luo, M. Effect of the regulation of retinoid X receptor-α gene expression on rat hepatic fibrosis. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2011, 41, 475–483. [Google Scholar] [CrossRef]
- Ram, P.T.; Iyengar, R. G protein coupled receptor signaling through the Src and Stat3 pathway: Role in proliferation and transformation. Oncogene 2001, 20, 1601–1606. [Google Scholar] [CrossRef]
- Bo, C.; Zhang, J.; Sai, L.; Du, Z.; Yu, G.; Li, C.; Li, M.; Peng, C.; Jia, Q.; Shao, H. Integrative transcriptomic and proteomic analysis reveals mechanisms of silica-induced pulmonary fibrosis in rats. BMC Pulm. Med. 2022, 22, 13. [Google Scholar] [CrossRef]
- Yao, J.; Liang, L.-H.; Zhang, Y.; Ding, J.; Tian, Q.; Li, J.-J.; He, X.-H. GNAI1 Suppresses Tumor Cell Migration and Invasion and is Post-Transcriptionally Regulated by Mir-320a/c/d in Hepatocellular Carcinoma. Cancer Biol. Med. 2012, 9, 234–241. [Google Scholar] [CrossRef]
- Haak, A.J.; Ducharme, M.T.; Diaz Espinosa, A.M.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharmacol. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sin. B 2022, 12, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Luo, J.; Cantley, L.C. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it’s implication in cancer. Cell Cycle (Georget. Tex.) 2005, 4, 1309–1312. [Google Scholar] [CrossRef]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Platé, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Man, Y.; Genovese, F.; et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead transcription factors: Key players in health and disease. Trends Genet. TIG 2011, 27, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Ma, Z.; Hu, W.; Jiang, S.; Yang, Z.; Li, T.; Chen, F.; Jia, G.; Yang, Y. FOXO1/3: Potential suppressors of fibrosis. Ageing Res. Rev. 2018, 41, 42–52. [Google Scholar] [CrossRef]
- Al-Tamari, H.M.; Dabral, S.; Schmall, A.; Sarvari, P.; Ruppert, C.; Paik, J.; DePinho, R.A.; Grimminger, F.; Eickelberg, O.; Guenther, A.; et al. FoxO3 an important player in fibrogenesis and therapeutic target for idiopathic pulmonary fibrosis. EMBO Mol. Med. 2018, 10, 276–293. [Google Scholar] [CrossRef]
- Rao, P.; Pang, M.; Qiao, X.; Yu, H.; Wang, H.; Yang, Y.; Ren, X.; Hu, M.; Chen, T.; Cao, Q.; et al. Promotion of β-catenin/Foxo1 signaling ameliorates renal interstitial fibrosis. Lab. Investig. 2019, 99, 1689–1701. [Google Scholar] [CrossRef]
- Wu, B.; Rockel, J.S.; Lagares, D.; Kapoor, M. Ephrins and Eph Receptor Signaling in Tissue Repair and Fibrosis. Curr. Rheumatol. Rep. 2019, 21, 23. [Google Scholar] [CrossRef]
- Darling, T.K.; Lamb, T.J. Emerging Roles for Eph Receptors and Ephrin Ligands in Immunity. Front. Immunol. 2019, 10, 454075. [Google Scholar] [CrossRef] [PubMed]
- Nunan, R.; Campbell, J.; Mori, R.; Pitulescu, M.E.; Jiang, W.G.; Harding, K.G.; Adams, R.H.; Nobes, C.D.; Martin, P. Ephrin-Bs Drive Junctional Downregulation and Actin Stress Fiber Disassembly to Enable Wound Re-epithelialization. Cell Rep. 2015, 13, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Mimche, P.N.; Lee, C.M.; Mimche, S.M.; Thapa, M.; Grakoui, A.; Henkemeyer, M.; Lamb, T.J. EphB2 receptor tyrosine kinase promotes hepatic fibrogenesis in mice via activation of hepatic stellate cells. Sci. Rep. 2018, 8, 2532. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, S.; Tang, A.; Al-Rabadi, L.; Henkemeyer, M.; Mimche, P.N.; Huang, Y. Key role for EphB2 receptor in kidney fibrosis. Clin. Sci. 2021, 135, 2127–2142. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N. Common Pathways in IPF and Lung Cancer. In Idiopathic Pulmonary Fibrosis: Advances in Diagnostic Tools and Disease Management; Nakamura, H., Aoshiba, K., Eds.; Springer: Tokyo, Japan, 2016; pp. 217–247. [Google Scholar]
- Vancheri, C. Idiopathic pulmonary fibrosis and cancer: Do they really look similar? BMC Med. 2015, 13, 220. [Google Scholar] [CrossRef]
- Gokey, J.J.; Snowball, J.; Sridharan, A.; Speth, J.P.; Black, K.E.; Hariri, L.P.; Perl, A.T.; Xu, Y.; Whitsett, J.A. MEG3 is increased in idiopathic pulmonary fibrosis and regulates epithelial cell differentiation. JCI Insight 2018, 3, e122490. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Liu, H.; Zhou, J.; Zhang, Q.; Bi, H. The MIR100HG/miR-29a-3p/Tab1 axis modulates TGF-β1-induced fibrotic changes in type II alveolar epithelial cells BLM-caused lung fibrogenesis in mice. Toxicol. Lett. 2022, 363, 45–54. [Google Scholar] [CrossRef]
- Roy, S.K.; Shrivastava, A.; Srivastav, S.; Shankar, S.; Srivastava, R.K. SATB2 is a novel biomarker and therapeutic target for cancer. J. Cell. Mol. Med. 2020, 24, 11064–11069. [Google Scholar] [CrossRef]
- Prakash, Y.; Thompson, M.A.; Meuchel, L.; Pabelick, C.M.; Mantilla, C.B.; Zaidi, S.; Martin, R.J. Neurotrophins in lung health and disease. Expert. Rev. Respir. Med. 2010, 4, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, A.R.; Buiting, C.; Kuiper, R.A.; Coolen, M.W. Regulatory Roles for Long ncRNA and mRNA. Cancers 2013, 5, 462–490. [Google Scholar] [CrossRef] [PubMed]
- Sebastian-delaCruz, M.; Gonzalez-Moro, I.; Olazagoitia-Garmendia, A.; Castellanos-Rubio, A.; Santin, I. The Role of lncRNAs in Gene Expression Regulation through mRNA Stabilization. Non-Coding RNA 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Assad, N.; Sood, A.; Campen, M.J.; Zychowski, K.E. Metal-Induced Pulmonary Fibrosis. Curr. Environ. Health Rep. 2018, 5, 486–498. [Google Scholar] [CrossRef]
- Schwaiblmair, M.; Behr, W.; Haeckel, T.; Märkl, B.; Foerg, W.; Berghaus, T. Drug induced interstitial lung disease. Open Respir. Med. J. 2012, 6, 63–74. [Google Scholar] [CrossRef]
- Brereton, C.J.; Jo, H.E. Acute exacerbations of idiopathic pulmonary fibrosis and the role of corticosteroids. Breathe 2020, 16, 200086. [Google Scholar] [CrossRef]
- Slattery, C.; Campbell, E.; McMorrow, T.; Ryan, M.P. Cyclosporine A-induced renal fibrosis: A role for epithelial-mesenchymal transition. Am. J. Pathol. 2005, 167, 395–407. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Azuma, A.; Inase, N.; Taniguchi, H.; Ogura, T.; Inoue, E.; Takeuchi, M.; Yoshizawa, Y.; Sugiyama, Y.; Kudoh, S. Cyclosporine A combined with low-dose corticosteroid treatment in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2015, 53, 288–295. [Google Scholar] [CrossRef]
- Kayhan, S.; Guzel, A.; Duran, L.; Tutuncu, S.; Guzel, A.; Gunaydın, M.; Salis, O.; Okuyucu, A.; Selcuk, M.Y. Effects of leflunomide on inflamation and fibrosis in bleomycine induced pulmonary fibrosis in wistar albino rats. J. Thorac. Dis. 2013, 5, 641–649. [Google Scholar] [CrossRef]
- El-Sherbiny, M.; Atef, H.; Eladl, M.A.; Mohamed, A.S.; El-Shafey, M.; Ali, H.S.; Zaitone, S.A.; Alomar, S.Y.; Alqahtani, S.A.M.; Aloyouni, S.Y.; et al. Leflunomide Induces Dose-Dependent Lung Injury in Mice via Stimulating Vimentin and NLRP3 Inflammasome Production. Front. Pharmacol. 2021, 12, 631216. [Google Scholar] [CrossRef]
- Matthew, J.P.; Joanne, E.M.; Patrick, M.B.; Isabelle, B.; Tammy, C.H.; Cynthia, D.M.; Larissa, S.; Jennifer, M.T.; Elie, A.A.; Sue, E.B.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | ID_GEO | Platform | Sample Size | |
|---|---|---|---|---|
| IPF | Control | |||
| Study_1 | GSE99621 | Illumina HiSeq 2500 | 18 | 8 |
| Study_2 | GSE180415 | Illumina HiSeq 4000 | 5 | 4 |
| Study_3 | GSE185492 | Illumina NovaSeq 6000 | 12 | 12 |
| Total sample size | 35 | 24 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Martínez, A.; Santos-Álvarez, J.C.; Velázquez-Enríquez, J.M.; Ramírez-Hernández, A.A.; Vásquez-Garzón, V.R.; Baltierrez-Hoyos, R. lncRNA-mRNA Co-Expression and Regulation Analysis in Lung Fibroblasts from Idiopathic Pulmonary Fibrosis. Non-Coding RNA 2024, 10, 26. https://doi.org/10.3390/ncrna10020026
López-Martínez A, Santos-Álvarez JC, Velázquez-Enríquez JM, Ramírez-Hernández AA, Vásquez-Garzón VR, Baltierrez-Hoyos R. lncRNA-mRNA Co-Expression and Regulation Analysis in Lung Fibroblasts from Idiopathic Pulmonary Fibrosis. Non-Coding RNA. 2024; 10(2):26. https://doi.org/10.3390/ncrna10020026
Chicago/Turabian StyleLópez-Martínez, Armando, Jovito Cesar Santos-Álvarez, Juan Manuel Velázquez-Enríquez, Alma Aurora Ramírez-Hernández, Verónica Rocío Vásquez-Garzón, and Rafael Baltierrez-Hoyos. 2024. "lncRNA-mRNA Co-Expression and Regulation Analysis in Lung Fibroblasts from Idiopathic Pulmonary Fibrosis" Non-Coding RNA 10, no. 2: 26. https://doi.org/10.3390/ncrna10020026