
Changes of the tRNA Modification Pattern during the Development of Dictyostelium discoideum

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
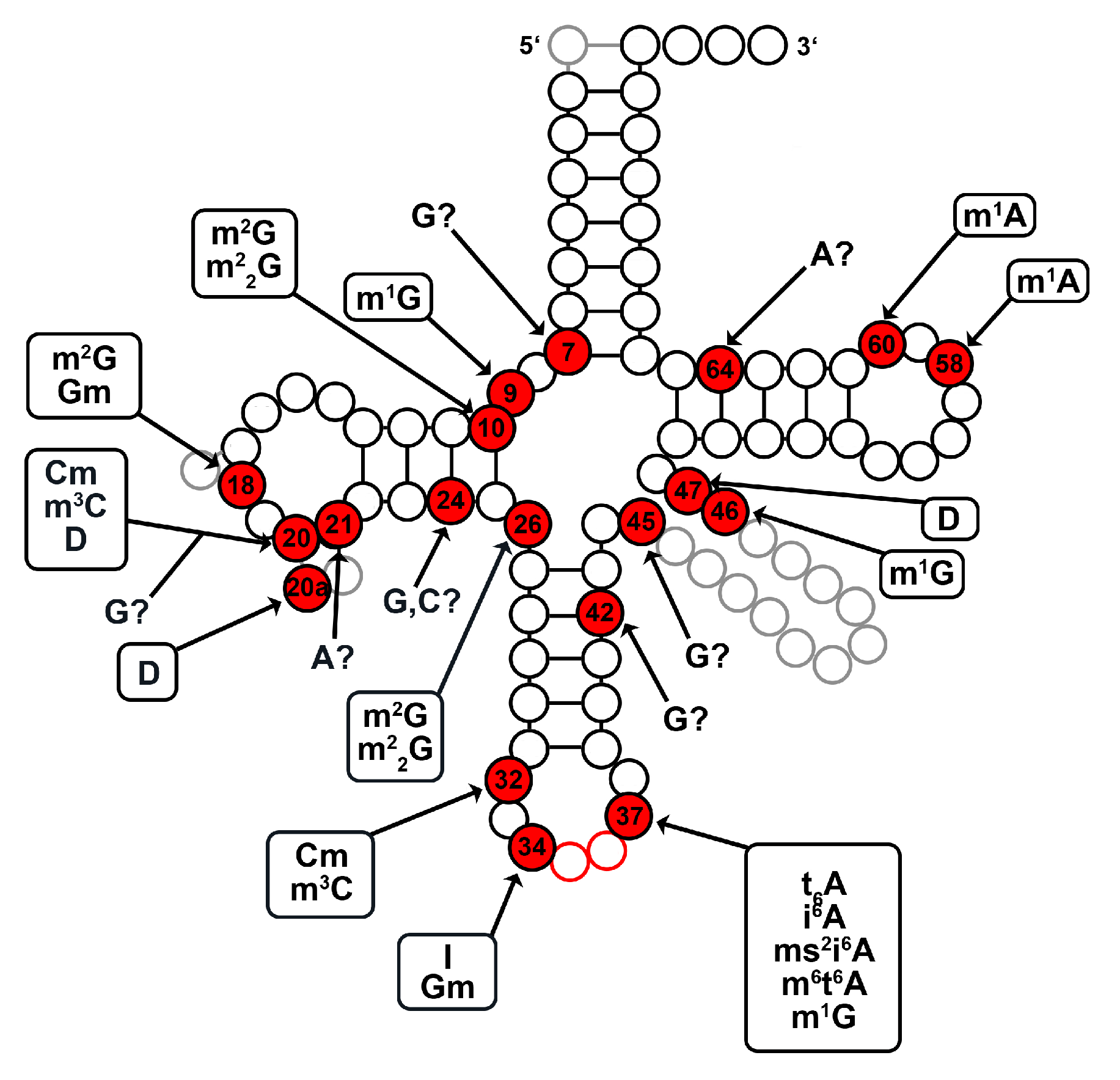
2.1. tRNA Modification Diversity in D. discoideum
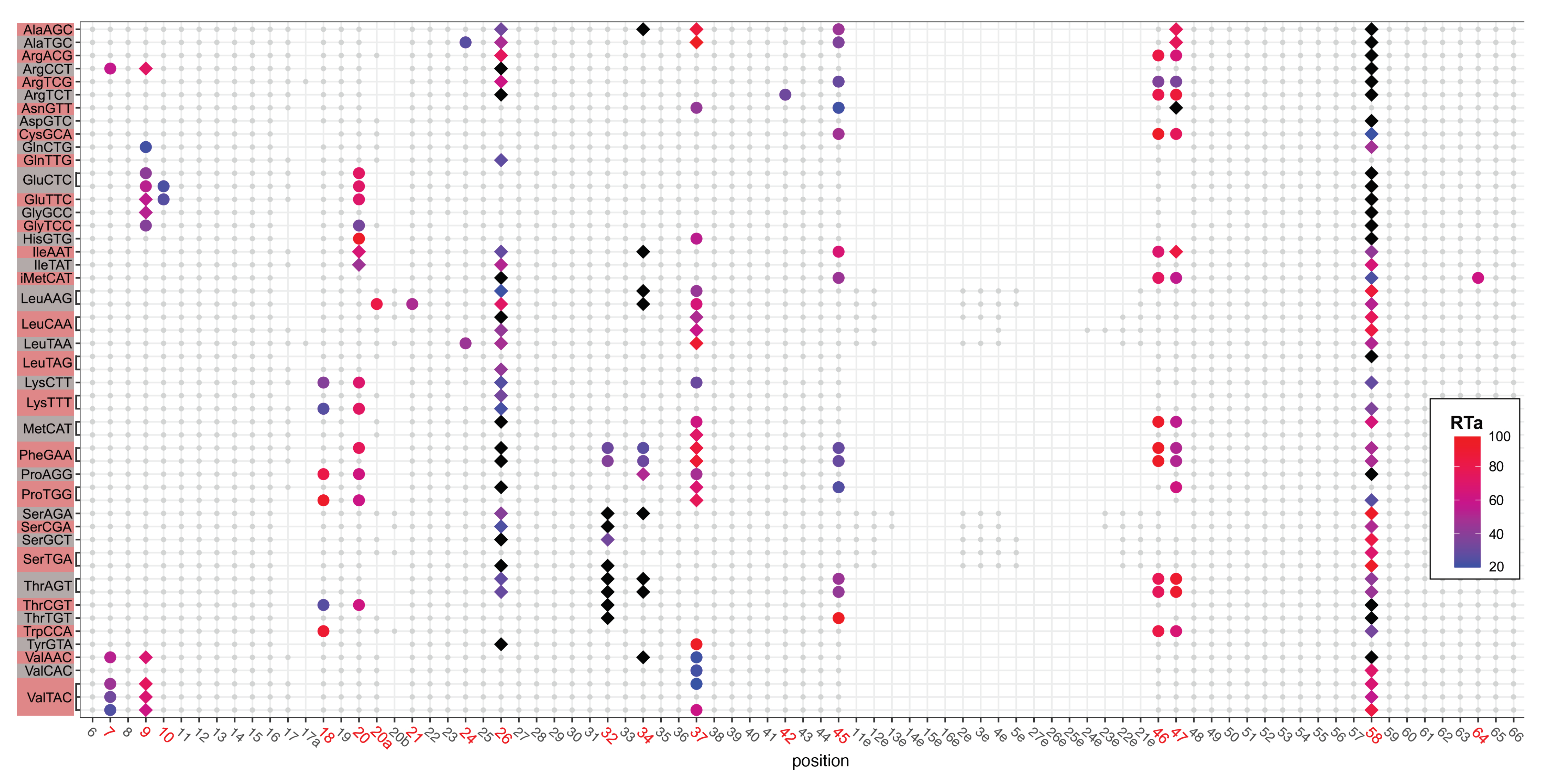
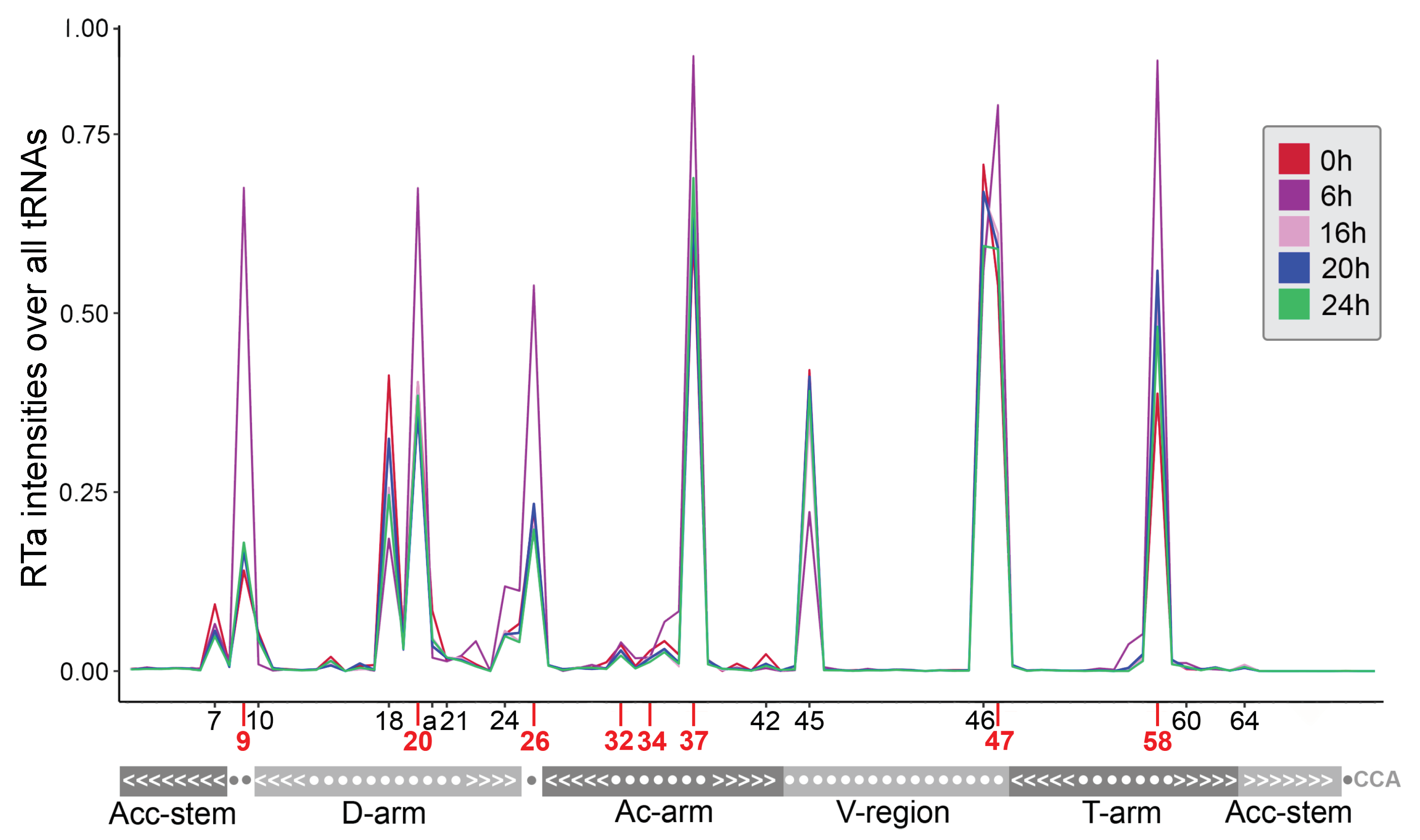
2.2. tRNA Modifications Vary during the D. discoideum Life Cycle
3. Discussion
4. Materials and Methods
4.1. Isolation of D. discoideum Total RNA
4.2. tRNA Read Mapping
4.3. Profiling of Modifications Sites in tRNAs
4.4. Fitting tRNAs to the Standard Model
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| cAMP | cyclic adenosine monophosphate |
| 2′-O-methylcytidine | |
| dihydrouridine | |
| ESA | enhanced suffix array |
| 2′-O-methylguanosine | |
| HAMR | high-throughput annotation of modified ribonucleotides |
| inosine | |
| -isopentenyladenosine | |
| LOTTE-seq | long hairpin oligonucleotide based tRNA high-throughput sequencing |
| miRNA | microRNA |
| mt-tRNA | mitochondrial tRNA |
| 2-methylthio--isopentenyladenosine | |
| 1-methyladenosine | |
| 1-methylguanosine | |
| -methylguanosine | |
| -dimethylguanosine | |
| 3-methylcytidine | |
| 5-methylcytidine | |
| 7-methylguanosine | |
| -methyl--threonylcarbamoyladenosine | |
| NCBI | Database resources of the National Center for Biotechnology Information |
| PCA | polymerase chain reaction |
| pre-tRNAs | precursor tRNAs |
| queuine | |
| rmRNA | ribo-minus RNA-seq |
| RNA-seq | RNA sequencing |
| RT | reverse transcription |
| RTa | reverse transcription arrest |
| -threonylcarbamoyladenosine | |
| snoRNA | small nucleolar RNA |
| tRNA | transfer RNA |
| pseudouridine |
References
- Sprinzl, M.; Cramer, F. The -C-C-A End of tRNA and Its Role in Protein Biosynthesis. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Ed.; Academic Press: Cambridge, MA, USA, 1979; Volume 22, pp. 1–69. [Google Scholar] [CrossRef]
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Olchowik, A.; Grosjean, H.; Bujnicki, J.M. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014, 11, 1619–1629. [Google Scholar] [CrossRef]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Suzuki, T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014, 42, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef]
- Duechler, M.; Leszczyńska, G.; Sochacka, E.; Nawrot, B. Nucleoside modifications in the regulation of gene expression: Focus on tRNA. Cell. Mol. Life Sci. 2016, 73, 3075–3095. [Google Scholar] [CrossRef]
- Nakai, Y.; Horiguchi, G.; Iwabuchi, K.; Harada, A.; Nakai, M.; Hara-Nishimura, I.; Yano, T. tRNA Wobble Modification Affects Leaf Cell Development in Arabidopsis thaliana. Plant Cell Physiol. 2019, 60, 2026–2039. [Google Scholar] [CrossRef]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Motorin, Y. Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol. 2017, 14, 1124–1137. [Google Scholar] [CrossRef]
- Vandivier, L.E.; Anderson, Z.D.; Gregory, B.D. HAMR High-Throughput Annotation of Modified Ribonucleotides. In Epitranscriptomics; Methods Mol. Biol. Volume 1870; Wajapeyee, N., Gupta, R., Clifton, N.J., Eds.; Springer: Berlin, Germany, 2019; pp. 51–67. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of Modified Residues in RNAs by Reverse Transcription-Based Methods. In RNA Modification; Methods in Enzymology Volume 425; Gott, J.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 21–53. [Google Scholar] [CrossRef]
- Findeiß, S.; Langenberger, D.; Stadler, P.F.; Hoffmann, S. Traces of Post-Transcriptional RNA Modifications in Deep Sequencing Data. Biol. Chem. 2011, 392, 305–313. [Google Scholar] [CrossRef]
- Ebhardt, H.A.; Tsang, H.H.; Dai, D.C.; Liu, Y.; Bostan, B.; Fahlman, R.P. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009, 37, 2461–2470. [Google Scholar] [CrossRef]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.S.W. HAMR: High-throughput annotation of modified ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thuring, K.; Winz, M.L.; Sharma, S.; Entian, K.D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Helm, M.; Motorin, Y. Use of specific chemical reagents for detection of modified nucleotides in RNA. J. Nucleic Acids 2011, 2011, 408053. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Cui, P.; Lin, Q.; Ding, F.; Xin, C.; Gong, W.; Zhang, L.; Geng, J.; Zhang, B.; Yu, X.; Yang, J.; et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010, 96, 259–265. [Google Scholar] [CrossRef]
- Erber, L.; Hoffmann, A.; Fallmann, J.; Betat, H.; Stadler, P.F.; Mörl, M. LOTTE-seq (Long hairpin oligonucleotide based tRNA high-throughput sequencing): Specific selection of tRNAs with 3′-CCA end for high-throughput sequencing. RNA Biol. 2020, 17, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006, 2, e20221. [Google Scholar] [CrossRef]
- Behrens, A.; Rodschinka, G.; Nedialkova, D.D. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell 2021, 81, 1802–1815.e7. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Stadler, P.F.; Strimmer, K. A Simple Data-Adaptive Probabilistic Variant Calling Model. Algorithms Mol. Biol. 2015, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Fallmann, J.; Vilardo, E.; Mörl, M.; Stadler, P.F.; Amman, F. Accurate Mapping of tRNA Reads. Bioinformatics 2018, 34, 1116–1124, Correction: Bioinformatics 2018, 34, 2359. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Werner, S.; Kemmer, T.; Niebler, S.; Kristen, M.; Ayadi, L.; Johe, P.; Marchand, V.; Schirmeister, T.; Motorin, Y.; et al. Graphical Workflow System for Modification Calling by Machine Learning of Reverse Transcription Signatures. Front. Genet. 2019, 10, 876. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Pseudo-Seq: Genome-Wide Detection of Pseudouridine Modifications in RNA. Methods Enzymol. 2015, 560, 219–245. [Google Scholar] [CrossRef]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m7G and m3C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Wintermeyer, W.; Zachau, H.G. A specific chemical chain scission of tRNA at 7-methylguanosine. FEBS Lett. 1970, 11, 160–164. [Google Scholar] [CrossRef]
- Squires, J.E.; Preiss, T. Function and detection of 5-methylcytosine in eukaryotic RNA. Epigenomics 2010, 2, 709–715. [Google Scholar] [CrossRef]
- Silberberg, G.; Öhman, M. The edited transcriptome: Novel high throughput approaches to detect nucleotide deamination. Curr. Opin. Genet. Dev. 2011, 21, 401–406. [Google Scholar] [CrossRef]
- Thomassin, H.; Oakeley, E.J.; Grange, T. Identification of 5-Methylcytosine in Complex Genomes. Methods 1999, 19, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toffano-Nioche, C.; Lorieux, F.; Gautheret, D.; Lehmann, J. Accurate characterization of Escherichia coli tRNA modifications with a simple method of deep-sequencing library preparation. RNA Biol. 2021, 18, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Dedon, P.C.; Waldor, M.K. Surveying the landscape of tRNA modifications by combining tRNA sequencing and RNA mass spectrometry. bioRxiv 2019, 723049. [Google Scholar] [CrossRef]
- Wolff, P.; Villette, C.; Zumsteg, J.; Heintz, D.; Antoine, L.; Chane-Woon-Ming, B.; Droogmans, L.; Grosjean, H.; Westhof, E. Comparative patterns of modified nucleotides in individual tRNA species from a mesophilic and two thermophilic archaea. RNA 2020, 26, 1957–1975. [Google Scholar] [CrossRef]
- Pinkard, O.; McFarland, S.; Sweet, T.; Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Comm. 2020, 11, 4104. [Google Scholar] [CrossRef]
- Marin, F.T. Regulation of development in Dictyostelium discoideum. Dev. Biol. 1976, 48, 110–117. [Google Scholar] [CrossRef]
- Loomis, W.F. Cell signaling during development of Dictyostelium. Dev. Biol. 2014, 391, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, R.L.; Firtel, R.A. Insights into morphogenesis from a simple developmental system. Nat. Rev. Mol. Cell Biol. 2004, 5, 531–541. [Google Scholar] [CrossRef] [PubMed]
- van Driessche, N.; Shaw, C.; Katoh, M.; Morio, T.; Sucgang, R.; Ibarra, M.; Kuwayama, H.; Saito, T.; Urushihara, H.; Maeda, M.; et al. A transcriptional profile of multicellular development in Dictyostelium discoideum. Development 2002, 129, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- González-Velasco, Ó.; de las Rivas, J.; Lacal, J. Proteomic and Transcriptomic Profiling Identifies Early Developmentally Regulated Proteins in Dictyostelium Discoideum. Cells 2019, 8, 1187. [Google Scholar] [CrossRef]
- Avesson, L.; Schumacher, H.T.; Fechter, P.; Romby, P.; Hellman, U.; Söderbom, F. Abundant class of non-coding RNA regulates development in the social amoeba Dictyostelium discoideum. RNA Biol. 2011, 8, 1094–1104. [Google Scholar] [CrossRef]
- Dingermann, T.; Schmidt, W.; Kersten, H. Modified bases in tRNA of Dictyostelium discoideum: Alterations in the ribothymidine content during development. FEBS Lett. 1977, 80, 205–208. [Google Scholar] [CrossRef]
- Dingermann, T.; Pistel, F.; Kersten, H. Early developmental changes in tRNA of Dictyostelium discoideum. Biochem. Soc. Trans. 1980, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Dingermann, T.; Pistel, F.; Kersten, H. Functional role of ribosylthymine in transfer RNA. Preferential utilization of tRNAs containing ribosylthymine instead of uridine at position 54 in protein synthesis of Dictyostelium discoideum. Eur. J. Biochem. 1980, 104, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Erber, L.; Hoffmann, A.; Fallmann, J.; Hagedorn, M.; Hammann, C.; Stadler, P.F.; Betat, H.; Prohaska, S.; Mörl, M. Unusual Occurrence of Two Bona-Fide CCA-Adding Enzymes in Dictyostelium discoideum. Int. J. Mol. Sci. 2020, 21, 5210. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, L.; Pachebat, J.A.; Glöckner, G.; Rajandream, M.A.; Sucgang, R.; Berriman, M.; Song, J.; Olsen, R.; Szafranski, K.; Xu, Q.; et al. The genome of the social amoeba Dictyostelium discoideum. Nature 2005, 435, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Marchand, V. Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update. Genes 2021, 12, 278. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.C.; Evans, M.E.; Dominissini, D.; Zheng, G.; Pan, T. tRNA base methylation identification and quantification via high-throughput sequencing. RNA 2016, 22, 1771–1784. [Google Scholar] [CrossRef]
- Sexton, A.N.; Wang, P.Y.; Rutenberg-Schoenberg, M.; Simon, M.D. Interpreting Reverse Transcriptase Termination and Mutation Events for Greater Insight into the Chemical Probing of RNA. Biochemistry 2017, 56, 4713–4721. [Google Scholar] [CrossRef]
- Xu, H.; Yao, J.; Wu, D.; Lambowitz, A.M. Improved TGIRT-seq methods for comprehensive transcriptome profiling with decreased adapter dimer formation and bias correction. Sci. Rep. 2019, 9, 7953. [Google Scholar] [CrossRef] [PubMed]
- Schäck, M.A.; Jablonski, K.P.; Gräf, S.; Klassen, R.; Schaffrath, R.; Kellner, S.; Hammann, C. Eukaryotic life without tQCUG: The role of Elongator-dependent tRNA modifications in Dictyostelium discoideum. Nucleic Acids Res. 2020, 48, 7899–7913. [Google Scholar] [CrossRef]
- Borland, K.; Diesend, J.; Ito-Kureha, T.; Heissmeyer, V.; Hammann, C.; Buck, A.H.; Michalakis, S.; Kellner, S. Production and Application of Stable Isotope-Labeled Internal Standards for RNA Modification Analysis. Genes 2019, 10, 26. [Google Scholar] [CrossRef]
- Sprinzl, M.; Horn, C.; Brown, M.; Ioudovitch, A.; Steinberg, S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998, 26, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.M.; Fay, M.M.; Ivanov, P. The role of RNA modifications in the regulation of tRNA cleavage. FEBS Lett. 2018, 592, 2828–2844. [Google Scholar] [CrossRef]
- Pang, Y.L.J.; Poruri, K.; Martinis, S.A. tRNA synthetase: tRNA aminoacylation and beyond. Wiley Int. Rev. RNA 2014, 5, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, D.; Gao, J.; Li, X.; Zhang, R.; Jin, X.; Hu, Z.; Zheng, B.; Persson, S.; Chen, P. The 2′-O-methyladenosine nucleoside modification gene OsTRM13 positively regulates salt stress tolerance in rice. J. Exp. Bot. 2017, 68, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Schachner, E.; Kersten, H. Queuine Deficiency and Restoration in Dictyostelium discoideum and Related Early Developmental Changes. Microbiology 1984, 130, 135–144. [Google Scholar] [CrossRef][Green Version]
- Rot, G.; Parikh, A.; Curk, T.; Kuspa, A.; Shaulsky, G.; Zupan, B. dictyExpress: A Dictyostelium discoideum gene expression database with an explorative data analysis web-based interface. BMC Bioinform. 2009, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Finer-Moore, J.; Czudnochowski, N.; O’Connell, J.D.; Wang, A.L.; Stroud, R.M. Crystal Structure of the Human tRNA m(1)A58 Methyltransferase-tRNA(3)(Lys) Complex: Refolding of Substrate tRNA Allows Access to the Methylation Target. J. Mol. Biol. 2015, 427, 3862–3876. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Windhof, I.M.; Maximov, V.; Jurkowski, T.; Jeltsch, A.; Förstner, K.U.; Sharma, C.M.; Gräf, R.; Nellen, W. Target recognition, RNA methylation activity and transcriptional regulation of the Dictyostelium discoideum Dnmt2-homologue (DnmA). Nucleic Acids Res. 2013, 41, 8615–8627. [Google Scholar] [CrossRef]
- Mutzel, R.; Malchow, D.; Meyer, D.; Kersten, H. tRNA (adenine-N1)-methyltransferase from Dictyostelium discoideum. Purification, characterization and developmental changes in activity. Eur. J. Biochem. 1986, 160, 101–108. [Google Scholar] [CrossRef]
- Chan, C.T.Y.; Dyavaiah, M.; DeMott, M.S.; Taghizadeh, K.; Dedon, P.C.; Begley, T.J. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010, 6, e1001247. [Google Scholar] [CrossRef]
- Chan, C.; Pham, P.; Dedon, P.C.; Begley, T.J. Lifestyle modifications: Coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol. 2018, 19, 228. [Google Scholar] [CrossRef]
- Pollo-Oliveira, L.; de Crécy-Lagard, V. Can Protein Expression Be Regulated by Modulation of tRNA Modification Profiles? Biochemistry 2019, 58, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ott, G.; Kersten, H.; Nishimura, S. Dictyostelium discoideum: A useful model system to evaluate the function of queuine and of the Q-family of tRNAs. FEBS Lett. 1982, 146, 311–314. [Google Scholar] [CrossRef]
- Torres, A.G.; Reina, O.; Stephan-Otto Attolini, C.; Ribas de Pouplana, L. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 8451–8456. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Andrews, S. FastQC, a Quality Control Tool for High Throughput Sequence Data. Available online: https://github.com/s-andrews/FastQC (accessed on 20 May 2021).
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinformatics 2014, 30, 1837–1843. [Google Scholar] [CrossRef]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comp. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Jühling, F.; Mörl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Pütz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Erber, L.; Betat, H.; Stadler, P.F.; Mörl, M.; Fallmann, J. Changes of the tRNA Modification Pattern during the Development of Dictyostelium discoideum. Non-Coding RNA 2021, 7, 32. https://doi.org/10.3390/ncrna7020032
Hoffmann A, Erber L, Betat H, Stadler PF, Mörl M, Fallmann J. Changes of the tRNA Modification Pattern during the Development of Dictyostelium discoideum. Non-Coding RNA. 2021; 7(2):32. https://doi.org/10.3390/ncrna7020032
Chicago/Turabian StyleHoffmann, Anne, Lieselotte Erber, Heike Betat, Peter F. Stadler, Mario Mörl, and Jörg Fallmann. 2021. "Changes of the tRNA Modification Pattern during the Development of Dictyostelium discoideum" Non-Coding RNA 7, no. 2: 32. https://doi.org/10.3390/ncrna7020032
APA StyleHoffmann, A., Erber, L., Betat, H., Stadler, P. F., Mörl, M., & Fallmann, J. (2021). Changes of the tRNA Modification Pattern during the Development of Dictyostelium discoideum. Non-Coding RNA, 7(2), 32. https://doi.org/10.3390/ncrna7020032