
MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome


Abstract
1. Introduction
2. Results
2.1. A Searchable 3′UTR Resource with miRNA Targets and 3′UTR Regulatory Motifs
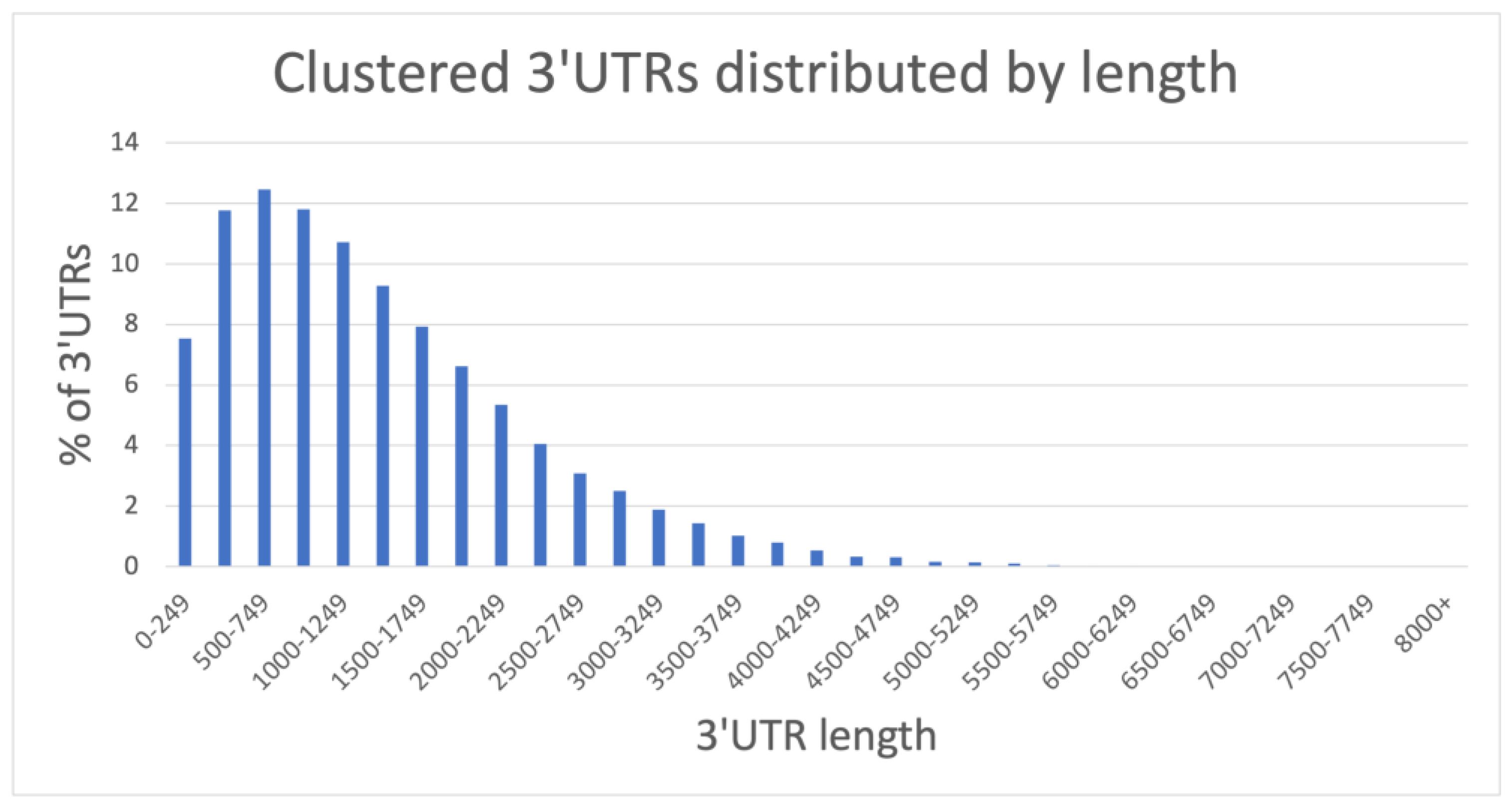
2.1.1. A Comprehensive 3′UTR Resource Extracted from FL-mRNAs
2.1.2. MicroSalmon: A Searchable Resource with In Silico Predicted miRNA Targets
2.1.3. The MicroSalmon Resource Also Includes Other Predicted Cis-Regulatory Motifs
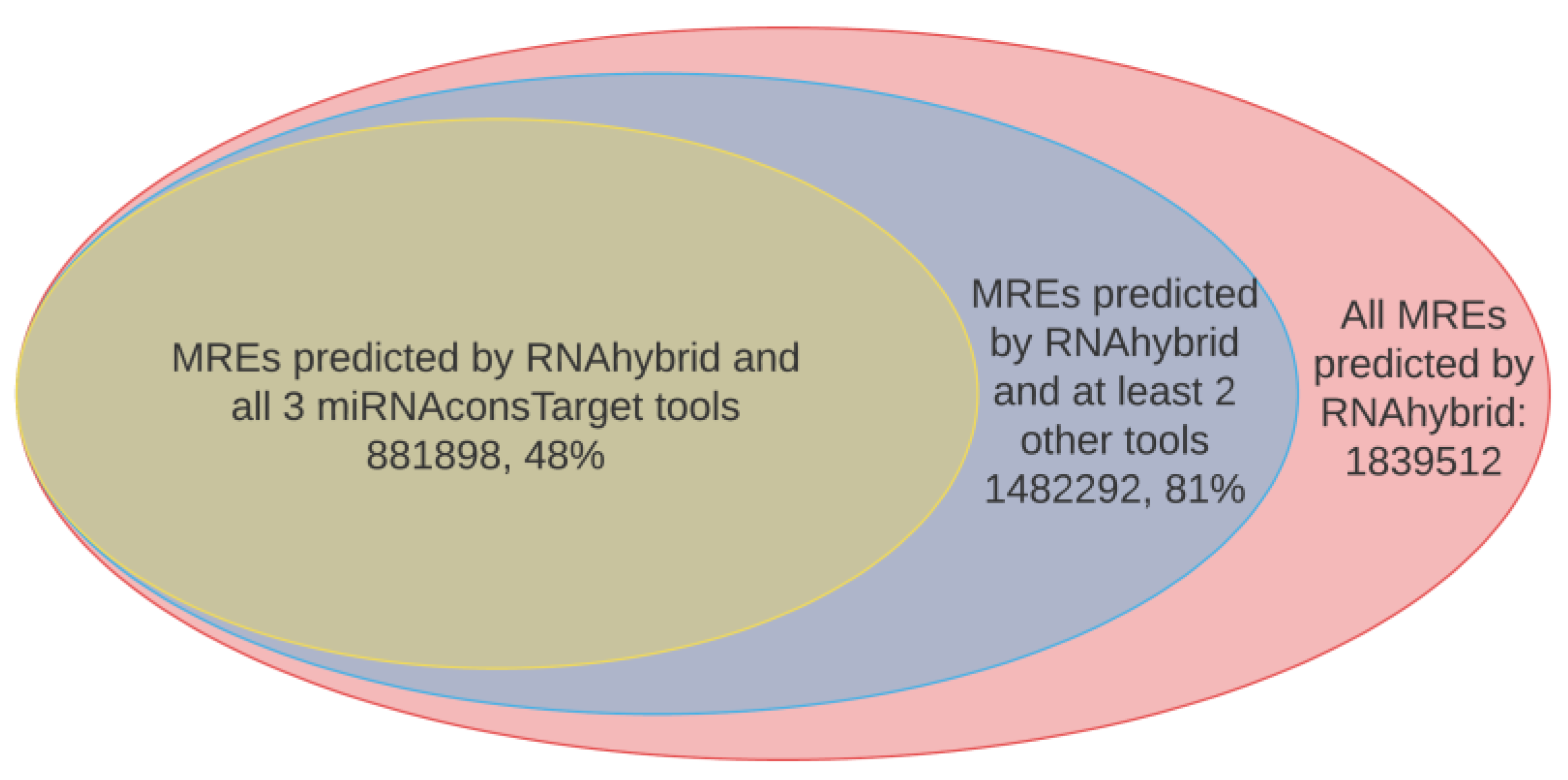
2.2. Results from In Silico miRNA Target Prediction Analysis
2.3. Identification of Known Cis-Regulatory Elements
2.4. Discovery of Novel Putative Functional Cis-Motifs by Their Over-Representation in the 3′UTRome
3. Discussion
3.1. The Accuracy and Limitations of In Silico miRNA Target Prediction
3.2. Identification of Known and Novel Cis-Regulatory Elements Greatly Expands the Knowledge of Transcript Regulation in Atlantic Salmon
3.3. The MicroSalmon Repistory
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. In Silico Prediction of miRNA Targets
4.2.2. Identification of Putative 3′UTR Cis-Regulatory Elements
4.2.3. The MicroSalmon GitHub Repository
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Tomari, Y. RISC assembly: Coordination between small RNAs and Argonaute proteins. Biochim. Biophys. Acta 2016, 1859, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Grillo, G.; Turi, A.; Licciulli, F.; Mignone, F.; Liuni, S.; Banfi, S.; Gennarino, V.A.; Horner, D.S.; Pavesi, G.; Picardi, E.; et al. UTRdb and UTRsite (RELEASE 2010): A collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010, 38, D75–D80. [Google Scholar] [CrossRef] [PubMed]
- Dassi, E.; Quattrone, A. Tuning the engine. RNA Biol. 2014, 9, 1224–1232. [Google Scholar] [CrossRef][Green Version]
- Szostak, E.; Gebauer, F. Translational control by 3′-UTR-binding proteins. Brief. Funct. Genom. 2012, 12, 58–65. [Google Scholar] [CrossRef]
- Andreassen, R.; Lunner, S.; Hoyheim, B. Characterization of full-length sequenced cDNA inserts (FLIcs) from Atlantic salmon (Salmo salar). BMC Genom. 2009, 10, 502. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Sturm, M.; Hackenberg, M.; Langenberger, D.; Frishman, D. TargetSpy: A supervised machine learning approach for microRNA target prediction. BMC Bioinform. 2010, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, R.; Hoyheim, B. miRNAs associated with immune response in teleost fish. Dev. Comp. Immunol. 2017, 75, 77–85. [Google Scholar] [CrossRef]
- Pinzon, N.; Li, B.; Martinez, L.; Sergeeva, A.; Presumey, J.; Apparailly, F.; Seitz, H. microRNA target prediction programs predict many false positives. Genome Res. 2017, 27, 234–245. [Google Scholar] [CrossRef]
- Woldemariam, N.T.; Agafonov, O.; Hoyheim, B.; Houston, R.D.; Taggart, J.B.; Andreassen, R. Expanding the miRNA Repertoire in Atlantic Salmon; Discovery of IsomiRs and miRNAs Highly Expressed in Different Tissues and Developmental Stages. Cells 2019, 8, 42. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, R.; Worren, M.M.; Hoyheim, B. Discovery and characterization of miRNA genes in Atlantic salmon (Salmo salar) by use of a deep sequencing approach. BMC Genom. 2013, 14, 482. [Google Scholar] [CrossRef]
- NCBI. NCBI Salmo salar Annotation Release 100 Assemblies Report. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Salmo_salar/100/ (accessed on 26 June 2020).
- Ramberg, S.; Høyheim, B.; Østbye, T.-K.K.; Andreassen, R. A de novo Full-Length mRNA Transcriptome Generated from Hybrid-Corrected PacBio Long-Reads Improves the Transcript Annotation and Identifies Thousands of Novel Splice Variants in Atlantic Salmon. Front. Genet. 2021, 12, 614. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, T.D.; Nome, T.; To, T.H.; Gundappa, M.K.; Macqueen, D.J.; Vage, D.I.; Sandve, S.R.; Hvidsten, T.R. SalMotifDB: A tool for analyzing putative transcription factor binding sites in salmonid genomes. BMC Genom. 2019, 20, 694. [Google Scholar] [CrossRef] [PubMed]
- Woldemariam, N.T.; Agafonov, O.; Sindre, H.; Hoyheim, B.; Houston, R.D.; Robledo, D.; Bron, J.E.; Andreassen, R. miRNAs Predicted to Regulate Host Anti-viral Gene Pathways in IPNV-Challenged Atlantic Salmon Fry Are Affected by Viral Load, and Associated With the Major IPN Resistance QTL Genotypes in Late Infection. Front. Immunol. 2020, 11, 2113. [Google Scholar] [CrossRef]
- Andreassen, R.; Woldemariam, N.T.; Egeland, I.O.; Agafonov, O.; Sindre, H.; Hoyheim, B. Identification of differentially expressed Atlantic salmon miRNAs responding to salmonid alphavirus (SAV) infection. BMC Genom. 2017, 18, 349. [Google Scholar] [CrossRef]
- Shwe, A.; Ostbye, T.K.; Krasnov, A.; Ramberg, S.; Andreassen, R. Characterization of Differentially Expressed miRNAs and Their Predicted Target Transcripts during Smoltification and Adaptation to Seawater in Head Kidney of Atlantic Salmon. Genes 2020, 11, 1059. [Google Scholar] [CrossRef] [PubMed]
- Østbye, T.K.K.; Woldemariam, N.T.; Lundberg, C.E.; Berge, G.M.; Ruyter, B.; Andreassen, R. Modulation of hepatic miRNA expression in Atlantic salmon (Salmo salar) by family background and dietary fatty acid composition. J. Fish Biol. 2021, 98, 1172–1185. [Google Scholar] [CrossRef]
- Williams, A.S.; Marzluff, W.F. The sequence of the stem and flanking sequences at the 3′ end of histone mRNA are critical determinants for the binding of the stem-loop binding protein. Nucleic Acids Res. 1995, 23, 654–662. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Galy, B.; Dandekar, T.; Bengert, P.; Vainshtein, Y.; Stolte, J.; Muckenthaler, M.U.; Hentze, M.W. Iron regulation and the cell cycle: Identification of an iron-responsive element in the 3′-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J. Biol. Chem. 2006, 281, 22865–22874. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef]
- Walczak, R.; Westhof, E.; Carbon, P.; Krol, A. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. RNA 1996, 2, 367–379. [Google Scholar] [PubMed]
- Walczak, R.; Carbon, P.; Krol, A. An essential non-Watson-Crick base pair motif in 3′UTR to mediate selenoprotein translation. RNA 1998, 4, 74–84. [Google Scholar]
- Fagegaltier, D.; Lescure, A.; Walczak, R.; Carbon, P.; Krol, A. Structural analysis of new local features in SECIS RNA hairpins. Nucleic Acids Res. 2000, 28, 2679–2689. [Google Scholar] [CrossRef]
- Korotkov, K.V.; Novoselov, S.V.; Hatfield, D.L.; Gladyshev, V.N. Mammalian selenoprotein in which selenocysteine (Sec) incorporation is supported by a new form of Sec insertion sequence element. Mol. Cell Biol. 2002, 22, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, S.V.; Rao, M.; Onoshko, N.V.; Zhi, H.; Kryukov, G.V.; Xiang, Y.; Weeks, D.P.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins and selenocysteine insertion system in the model plant cell system, Chlamydomonas reinhardtii. EMBO J. 2002, 21, 3681–3693. [Google Scholar] [CrossRef]
- Copeland, P.R.; Fletcher, J.E.; Carlson, B.A.; Hatfield, D.L.; Driscoll, D.M. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 2000, 19, 306–314. [Google Scholar] [CrossRef]
- Lescure, A.; Fagegaltier, D.; Carbon, P.; Krol, A. Protein factors mediating selenoprotein synthesis. Curr. Protein Pept. Sci. 2002, 3, 143–151. [Google Scholar] [CrossRef]
- Allmang, C.; Carbon, P.; Krol, A. The SBP2 and 15.5 kD/Snu13p proteins share the same RNA binding domain: Identification of SBP2 amino acids important to SECIS RNA binding. RNA 2002, 8, 1308–1318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chavatte, L.; Brown, B.A.; Driscoll, D.M. Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 2005, 12, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Lescure, A.; Allmang, C.; Yamada, K.; Carbon, P.; Krol, A. cDNA cloning, expression pattern and RNA binding analysis of human selenocysteine insertion sequence (SECIS) binding protein 2. Gene 2002, 291, 279–285. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Tujebajeva, R.M.; Copeland, P.R.; Xu, X.M.; Carlson, B.A.; Harney, J.W.; Driscoll, D.M.; Hatfield, D.L.; Berry, M.J. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000, 1, 158–163. [Google Scholar] [CrossRef]
- Grundner-Culemann, E.; Martin, G.W., 3rd; Harney, J.W.; Berry, M.J. Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes. RNA 1999, 5, 625–635. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vassalli, J.D.; Stutz, A. Translational control. Awakening dormant mRNAs. Curr. Biol. 1995, 5, 476–479. [Google Scholar] [CrossRef]
- Wickens, M.; Goodwin, E.B.; Kimble, J.; Strickland, S.; Hentze, M. Translational control of developmental decisions. Cold Spring Harb. Monogr. Ser. 2000, 39, 295–370. [Google Scholar]
- Verrotti, A.C.; Thompson, S.R.; Wreden, C.; Strickland, S.; Wickens, M. Evolutionary conservation of sequence elements controlling cytoplasmic polyadenylylation. Proc. Natl. Acad. Sci. USA 1996, 93, 9027–9032. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.B.; Okkema, P.G.; Evans, T.C.; Kimble, J. Translational regulation of tra-2 by its 3′ untranslated region controls sexual identity in C. elegans. Cell 1993, 75, 329–339. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H.; Standart, N.; Thiele, B.J. Translation of 15-lipoxygenase mRNA is inhibited by a protein that binds to a repeated sequence in the 3′ untranslated region. EMBO J. 1994, 13, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Ostareck, D.H.; Ostareck-Lederer, A.; Wilm, M.; Thiele, B.J.; Mann, M.; Hentze, M.W. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell 1997, 89, 597–606. [Google Scholar] [CrossRef]
- Ostareck-Lederer, A.; Ostareck, D.H.; Hentze, M.W. Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem. Sci. 1998, 23, 409–411. [Google Scholar] [CrossRef]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Boado, R.J.; Pardridge, W.M. Ten nucleotide cis element in the 3′-untranslated region of the GLUT1 glucose transporter mRNA increases gene expression via mRNA stabilization. Brain Res. Mol. Brain Res. 1998, 59, 109–113. [Google Scholar] [CrossRef]
- Banerjee, H.; Rahn, A.; Davis, W.; Singh, R. Sex lethal and U2 small nuclear ribonucleoprotein auxiliary factor (U2AF65) recognize polypyrimidine tracts using multiple modes of binding. RNA 2003, 9, 88–99. [Google Scholar] [CrossRef]
- Samuels, M.E.; Bopp, D.; Colvin, R.A.; Roscigno, R.F.; Garcia-Blanco, M.A.; Schedl, P. RNA binding by Sxl proteins in vitro and in vivo. Mol. Cell Biol. 1994, 14, 4975–4990. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.; Deshpande, G.; Schedl, P. Activities of the Sex-lethal protein in RNA binding and protein:protein interactions. Nucleic Acids Res. 1998, 26, 2625–2637. [Google Scholar] [CrossRef]
- Singh, R.; Valcarcel, J.; Green, M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 1995, 268, 1173–1176. [Google Scholar] [CrossRef]
- Wang, J.; Bell, L.R. The Sex-lethal amino terminus mediates cooperative interactions in RNA binding and is essential for splicing regulation. Genes Dev. 1994, 8, 2072–2085. [Google Scholar] [CrossRef]
- Boussadia, O.; Jacquemin-Sablon, H.; Dautry, F. Exon skipping in the expression of the gene immediately upstream of N-ras (unr/NRU). Biochim Biophys. Acta 1993, 1172, 64–72. [Google Scholar] [CrossRef]
- Boussadia, O.; Niepmann, M.; Creancier, L.; Prats, A.C.; Dautry, F.; Jacquemin-Sablon, H. Unr is required in vivo for efficient initiation of translation from the internal ribosome entry sites of both rhinovirus and poliovirus. J. Virol. 2003, 77, 3353–3359. [Google Scholar] [CrossRef]
- Chang, T.C.; Yamashita, A.; Chen, C.Y.; Yamashita, Y.; Zhu, W.; Durdan, S.; Kahvejian, A.; Sonenberg, N.; Shyu, A.B. UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant. Genes Dev. 2004, 18, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R.; Mitchell, S.A.; Spriggs, K.A.; Ostrowski, J.; Bomsztyk, K.; Ostarek, D.; Willis, A.E. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 2003, 22, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.L.; Hsuan, J.J.; Totty, N.; Jackson, R.J. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev 1999, 13, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin-Sablon, H.; Triqueneaux, G.; Deschamps, S.; le Maire, M.; Doniger, J.; Dautry, F. Nucleic acid binding and intracellular localization of unr, a protein with five cold shock domains. Nucleic Acids Res. 1994, 22, 2643–2650. [Google Scholar] [CrossRef]
- Jeffers, M.; Paciucci, R.; Pellicer, A. Characterization of unr; a gene closely linked to N-ras. Nucleic Acids Res. 1990, 18, 4891–4899. [Google Scholar]
- Mitchell, S.A.; Brown, E.C.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. Protein factor requirements of the Apaf-1 internal ribosome entry segment: Roles of polypyrimidine tract binding protein and upstream of N-ras. Mol. Cell Biol. 2001, 21, 3364–3374. [Google Scholar] [CrossRef]
- Mitchell, S.A.; Spriggs, K.A.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol. Cell 2003, 11, 757–771. [Google Scholar] [CrossRef]
- Tinton, S.A.; Schepens, B.; Bruynooghe, Y.; Beyaert, R.; Cornelis, S. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: Roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem. J. 2005, 385, 155–163. [Google Scholar] [CrossRef]
- Triqueneaux, G.; Velten, M.; Franzon, P.; Dautry, F.; Jacquemin-Sablon, H. RNA binding specificity of Unr, a protein with five cold shock domains. Nucleic Acids Res. 1999, 27, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Castagnetti, S.; Hentze, M.W.; Ephrussi, A.; Gebauer, F. Control of oskar mRNA translation by Bruno in a novel cell-free system from Drosophila ovaries. Development 2000, 127, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Kim-Ha, J.; Kerr, K.; Macdonald, P.M. Translational regulation of oskar mRNA by bruno, an ovarian RNA-binding protein, is essential. Cell 1995, 81, 403–412. [Google Scholar] [CrossRef]
- Parsch, J.; Russell, J.A.; Beerman, I.; Hartl, D.L.; Stephan, W. Deletion of a conserved regulatory element in the Drosophila Adh gene leads to increased alcohol dehydrogenase activity but also delays development. Genetics 2000, 156, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Parsch, J.; Stephan, W.; Tanda, S. A highly conserved sequence in the 3′-untranslated region of the drosophila Adh gene plays a functional role in Adh expression. Genetics 1999, 151, 667–674. [Google Scholar] [CrossRef]
- Lai, E.C.; Posakony, J.W. Regulation of Drosophila neurogenesis by RNA:RNA duplexes? Cell 1998, 93, 1103–1104. [Google Scholar] [CrossRef][Green Version]
- Lai, E.C.; Bodner, R.; Kavaler, J.; Freschi, G.; Posakony, J.W. Antagonism of notch signaling activity by members of a novel protein family encoded by the bearded and enhancer of split gene complexes. Development 2000, 127, 291–306. [Google Scholar] [CrossRef]
- Lai, E.C.; Burks, C.; Posakony, J.W. The K box, a conserved 3′ UTR sequence motif, negatively regulates accumulation of enhancer of split complex transcripts. Development 1998, 125, 4077–4088. [Google Scholar] [CrossRef]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Hew, Y.; Lau, C.; Grzelczak, Z.; Keeley, F.W. Identification of a GA-rich sequence as a protein-binding site in the 3′-untranslated region of chicken elastin mRNA with a potential role in the developmental regulation of elastin mRNA stability. J. Biol. Chem. 2000, 275, 24857–24864. [Google Scholar] [CrossRef]
- Tillmar, L.; Carlsson, C.; Welsh, N. Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J. Biol. Chem. 2002, 277, 1099–1106. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2016, 8, 6376–6398. [Google Scholar] [CrossRef]
- Shi, C.-Y.; Fan, Y.; Liu, B.; Lou, W.-H. HIF1 Contributes to Hypoxia-Induced Pancreatic Cancer Cells Invasion via Promoting QSOX1 Expression. Cell. Physiol. Biochem. 2013, 32, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Barreau, C. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef] [PubMed]
- Schelhorn, C.; Gordon, J.M.B.; Ruiz, L.; Alguacil, J.; Pedroso, E.; Macias, M.J. RNA recognition and self-association of CPEB4 is mediated by its tandem RRM domains. Nucleic Acids Res. 2014, 42, 10185–10195. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Abdel Messih, M.; Takacs, C.M.; Yartseva, V.; Oikonomou, P.; Christiano, R.; Stoeckius, M.; Lau, S.; Lee, M.T.; Beaudoin, J.-D.; et al. Genome wide analysis of 3′ UTR sequence elements and proteins regulating mRNA stability during maternal-to-zygotic transition in zebrafish. Genome Res. 2019, 29, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.; Shulman, E.D.; Elkon, R. Systematic identification of functional SNPs interrupting 3′UTR polyadenylation signals. PLOS Genet. 2020, 16. [Google Scholar] [CrossRef]
- Cheng, Y.; Miura, R.M.; Tian, B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics 2006, 22, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Gautheret, D. Sequence determinants in human polyadenylation site selection. BMC Genom. 2003, 4, 7. [Google Scholar] [CrossRef]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef]
- Riffo-Campos, A.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Bracken, C.P.; Goodall, G.J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011, 39, 6845–6853. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Lieberman, J.; Lal, A. Desperately seeking microRNA targets. Nat. Struct. Mol. Biol. 2010, 17, 1169–1174. [Google Scholar] [CrossRef]
- Elton, T.S.; Yalowich, J.C. Experimental procedures to identify and validate specific mRNA targets of miRNAs. EXCLI J. 2015, 14, 758–790. [Google Scholar] [CrossRef]
- Giuffra, E.; Tuggle, C.K.; Consortium, F. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef]
- Corà, D.; Di Cunto, F.; Caselle, M.; Provero, P. Identification of candidate regulatory sequences in mammalian 3′ UTRs by statistical analysis of oligonucleotide distributions. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoon, K.; Ko, D.; Doderer, M.; Livi, C.B.; Penalva, L.O. Over-represented sequences located on 3′ UTRs are potentially involved in regulatory functions. RNA Biol. 2008, 5, 255–262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andreassen, R.; Lundsted, J.; Olaisen, B. Mutation at minisatellite locus DYF155S1: Allele length mutation rate is affected by age of progenitor. Electrophoresis 2002, 23, 2377–2383. [Google Scholar] [CrossRef]
- Brinkmann, B.; Klintschar, M.; Neuhuber, F.; Hühne, J.; Rolf, B. Mutation Rate in Human Microsatellites: Influence of the Structure and Length of the Tandem Repeat. Am. J. Human Genet. 1998, 62, 1408–1415. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Mennigen, J.A.; Zhang, D. MicroTrout: A comprehensive, genome-wide miRNA target prediction framework for rainbow trout, Oncorhynchus mykiss. Comp. Biochem. Physiol. Part D Genom. Proteom. 2016, 20, 19–26. [Google Scholar] [CrossRef]
- Yang, J.; Liu, A.; He, I.; Bai, Y. Bioinformatics Analysis Revealed Novel 3′UTR Variants Associated with Intellectual Disability. Genes 2020, 11, 998. [Google Scholar] [CrossRef] [PubMed]
- Pichon, X.; A. Wilson, L.; Stoneley, M.; Bastide, A.; A. King, H.; Somers, J.; E. Willis, A.; A. King, H. RNA Binding Protein/RNA Element Interactions and the Control of Translation. Curr. Protein Peptide Sci. 2012, 13, 294–304. [Google Scholar] [CrossRef]
- Ali, A.; Thorgaard, G.H.; Salem, M. PacBio Iso-Seq Improves the Rainbow Trout Genome Annotation and Identifies Alternative Splicing Associated With Economically Important Phenotypes. Front. Genet. 2021, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Rueda, A.; Barturen, G.; Lebrón, R.; Gómez-Martín, C.; Alganza, Á.; Oliver, J.L.; Hackenberg, M. sRNAtoolbox: An integrated collection of small RNA research tools. Nucleic Acids Res. 2015, 43, W467–W473. [Google Scholar] [CrossRef] [PubMed]
- Rigoutsos, I.; Floratos, A. Combinatorial pattern discovery in biological sequences: The TEIRESIAS algorithm. Bioinformatics 1998, 14, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Orlov, Y.L.; Te Boekhorst, R.; Abnizova, I.I. Statistical measures of the structure of genomic sequences: Entropy, complexity, and position information. J. Bioinform. Comput. Biol. 2006, 4, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. Making sense of the human genome. Structure and Methods. In Proceedings of the 6th Conversation in the Discipline Biomolecular Stereodynamics Held at the State University of New York, Albany, NY, USA, 6–10 June 1989; Sarma, R.H., Samra, M.H., Eds.; Adenine Press: Schenectady, NY, USA, 1990. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UTRscan Identifier | Motif Name | Total Occurrences 1 | Unique Transcripts 2 | Different Loci 3 | Description | References |
|---|---|---|---|---|---|---|
| U0001 | HSL3 | 5 | 3 | 2 | Histone 3′UTR stem-loop structure | [30,31] |
| U0002 | IRE | 185 | 138 | 49 | Iron-responsive element | [32,33,34] |
| U0003 | SECIS1 | 861 | 857 | 294 | Selenocysteine insertion sequence—type 1 | [35,36,37,38,39,40,41,42,43,44,45,46,47] |
| U0004 | SECIS2 | 786 | 784 | 266 | Selenocysteine insertion sequence—type 2 | [35,36,37,38,39,40,41,42,43,44,45,46,47] |
| U0006 | CPE | 4685 | 4685 | 1633 | Cytoplasmic polyadenylation element | [48,49,50] |
| U0007 | TGE | 179 | 175 | 49 | TGE translational regulation element | [51] |
| U0009 | 15-LOX-DICE | 82 | 82 | 36 | 15-Lipoxygenase differentiation control element | [52,53,54] |
| U0010 | ARE2 | 108 | 108 | 49 | AU-rich class-2 element | [55] |
| U0012 | GLUT1 | 79 | 79 | 33 | Glusose transporter type-1 3′UTR cis-acting element | [56] |
| U0016 | SXL_BS | 6495 | 5941 | 2123 | SXL binding site | [57,58,59,60,61] |
| U0017 | UNR-bs | 5883 | 5420 | 2039 | UNR binding site | [62,63,64,65,66,67,68,69,70,71,72] |
| U0019 | BRE | 355 | 345 | 122 | Bruno 3′UTR responsive element | [73,74] |
| U0020 | ADH-DRE | 1529 | 1461 | 556 | Alcohol dehydrogenase 3′UTR downregulation control element | [75,76] |
| U0022 | PRONEURAL-BOX | 1 | 1 | 1 | Proneural box | [77,78] |
| U0023 | K-BOX | 17188 | 14341 | 5159 | K-box | [79,80] |
| U0024 | BRD-BOX | 7279 | 6635 | 2319 | Brd-box | [79,80] |
| U0025 | GY-BOX | 4990 | 4433 | 1693 | GY-box | [78,79,80] |
| U0027 | G3A | 11 | 11 | 3 | Elastin G3A 3′UTR stability motif | [81] |
| U0028 | INS_SCE | 14 | 14 | 6 | Insulin 3′UTR stability element | [82] |
| Iron-Relevant GO Terms 1 | Manual Gene Annotation 2 |
|---|---|
| Iron binding | timm2 |
| Iron ion homeostasis | meltf |
| Transferrin receptor | trfc paralog 1 |
| Transferrin receptor | trfc paralog 2 |
| —3 | qsox1 |
| Metalloreductase | steap3 |
| Iron ion binding | agmo |
| Heme binding | dgcr8 |
| Iron ion binding | fa2h |
| Motif length 1 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| All motifs 2 | 1 | 81 | 230 | 199 | 59 | 19 | 9 | 3 | 2 | 1 |
| Motifs containing a seed sequence 3 | 0 | 4 | 36 | 34 | 6 | 2 | 0 | 0 | 0 | 0 |
| Complexity filter motifs (CT > 0.27) 4 | 0 | 3 | 44 | 33 | 2 | 0 | 0 | 0 | 0 | 0 |
| Filtered motifs containing a seed sequence 5 | 0 | 1 | 9 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramberg, S.; Andreassen, R. MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome. Non-Coding RNA 2021, 7, 61. https://doi.org/10.3390/ncrna7040061
Ramberg S, Andreassen R. MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome. Non-Coding RNA. 2021; 7(4):61. https://doi.org/10.3390/ncrna7040061
Chicago/Turabian StyleRamberg, Sigmund, and Rune Andreassen. 2021. "MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome" Non-Coding RNA 7, no. 4: 61. https://doi.org/10.3390/ncrna7040061
APA StyleRamberg, S., & Andreassen, R. (2021). MicroSalmon: A Comprehensive, Searchable Resource of Predicted MicroRNA Targets and 3′UTR Cis-Regulatory Elements in the Full-Length Sequenced Atlantic Salmon Transcriptome. Non-Coding RNA, 7(4), 61. https://doi.org/10.3390/ncrna7040061