
A Universal Strategy for the Efficient Expression of Nanobodies in Pichia pastoris
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Medium
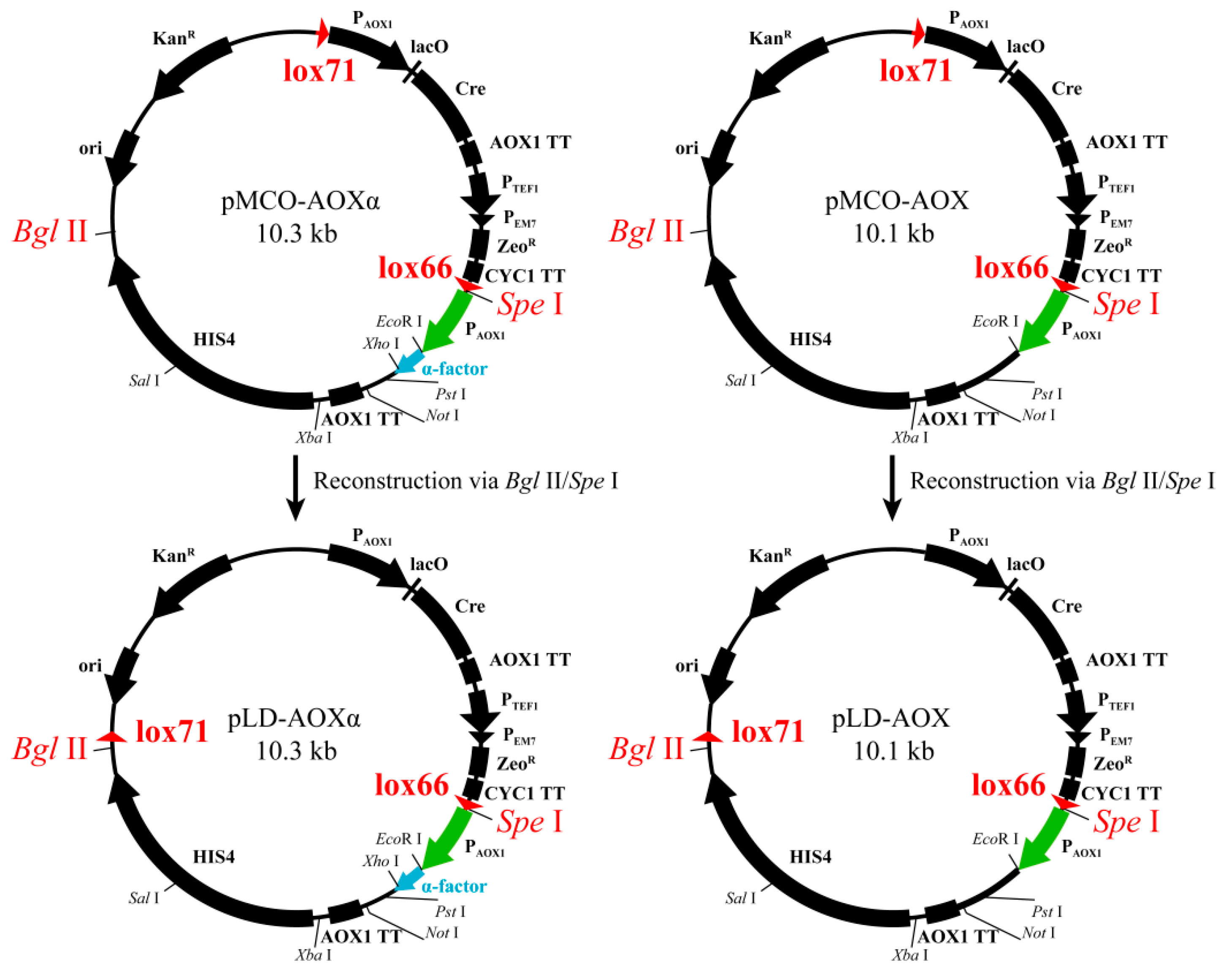
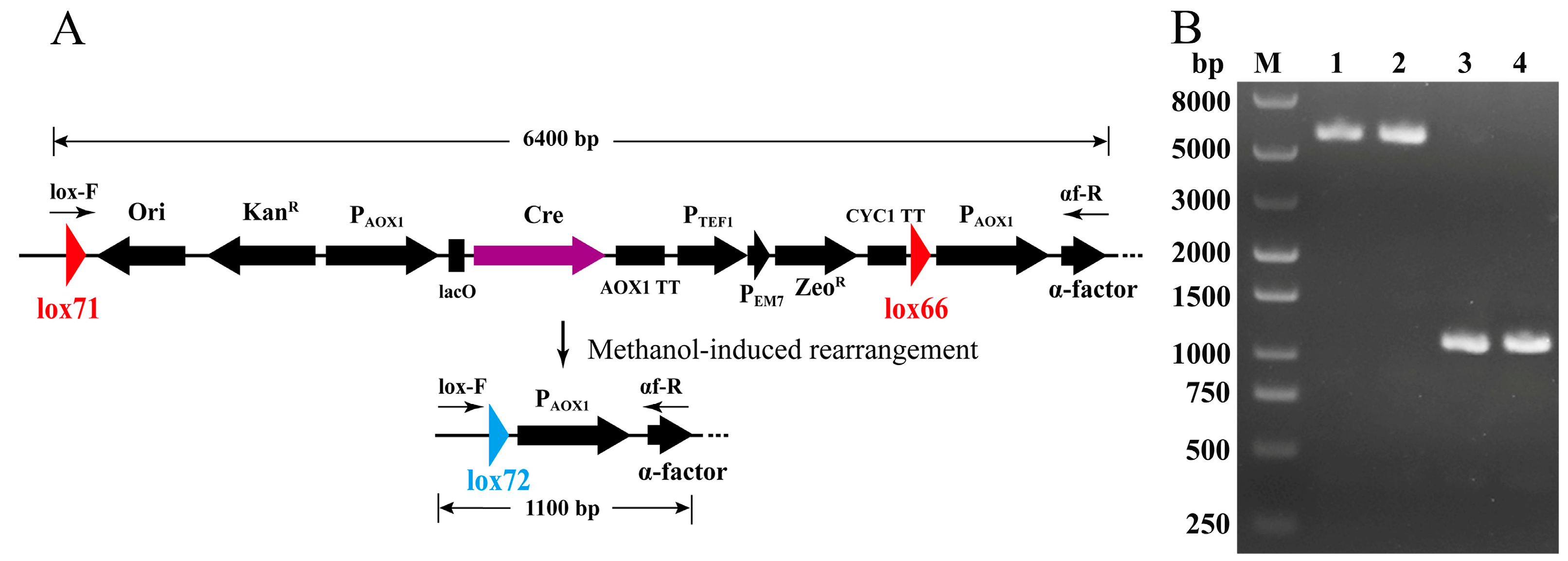
2.2. Construction of pLD-AOXα and pLD-AOX and Identification of Cre-loxP-Mediated Genetic Rearrangement
2.3. Construction of Plasmids with Alternative Promoters and Signal Peptides
2.4. Construction of Single-Copy Expression Plasmids and Yeast Transformants
2.5. Shake-Flask Culture and Semi-Quantitative Analysis
2.6. Construction of the Tandem Multicopy Expression Plasmids and Yeast Transformants
2.7. Obliteration of Resistance towards Zeocin for P. pastoris Transformants
2.8. Construction of Recombinant Plasmids with Various Molecular Chaperones
2.9. Generation of Multicopy Recombinants Co-Expressing Molecular Chaperones
2.10. High-Density Fermentation
2.11. Generation and Validation of Yeast Strains with Pre-Integrated Attenuated HAC1
2.12. Statistical Analysis
3. Results
3.1. Construction of pLD-AOXα and pLD-AOX and Identification of Cre-loxP Mediated Genetic Rearrangement
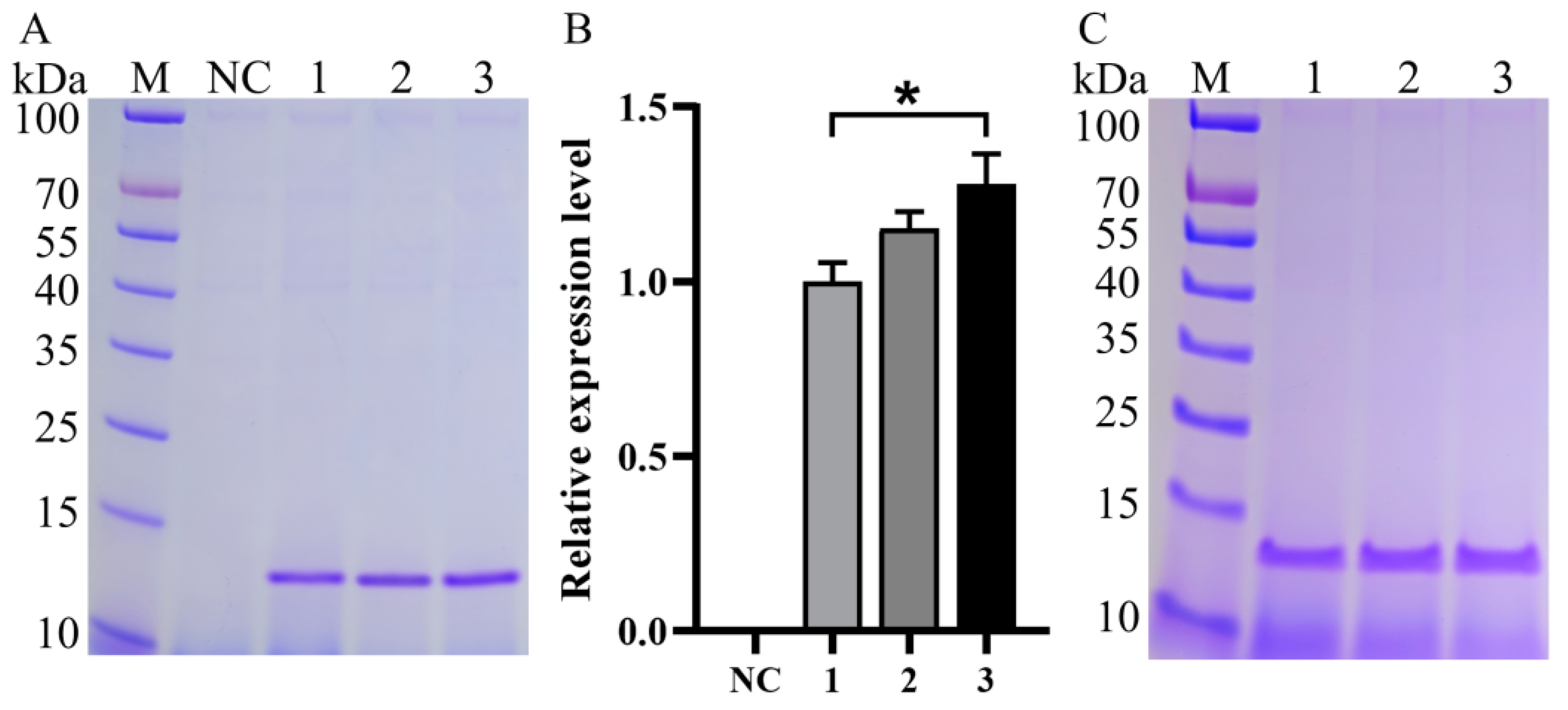
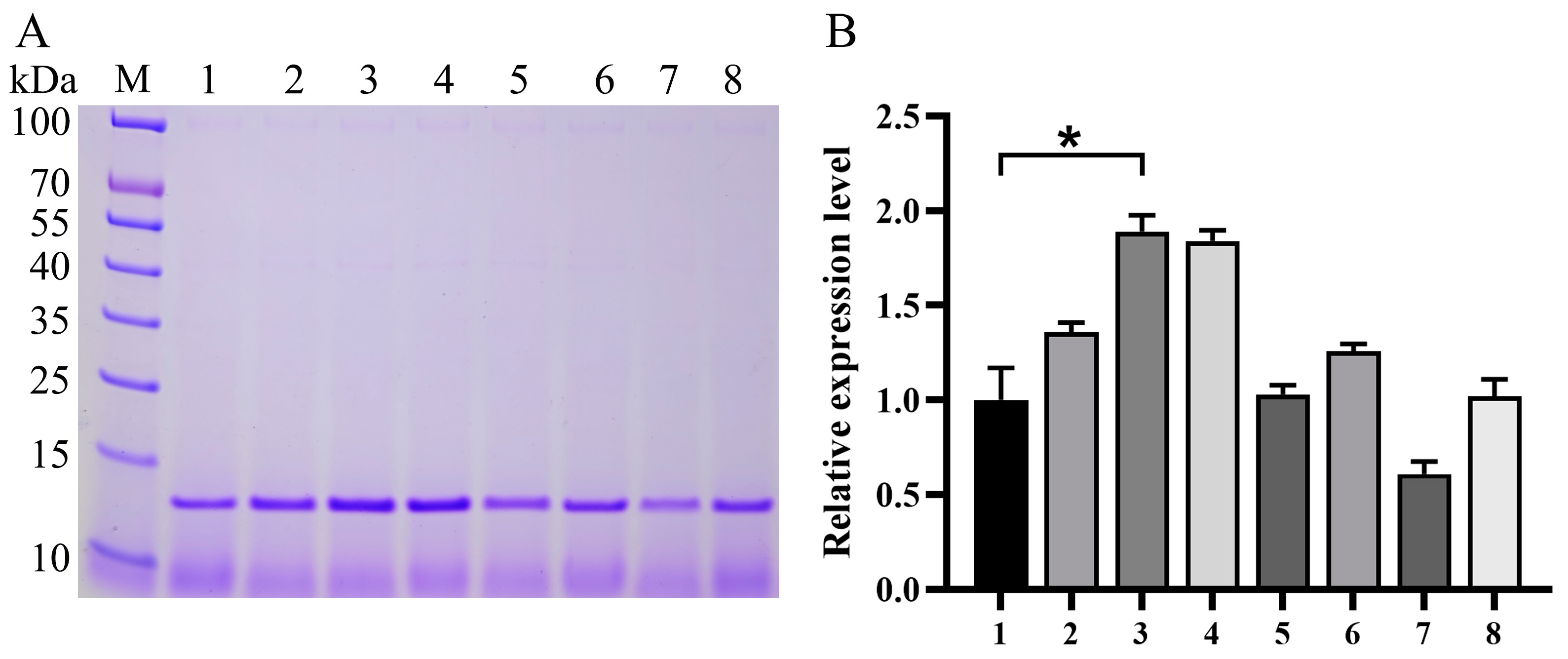
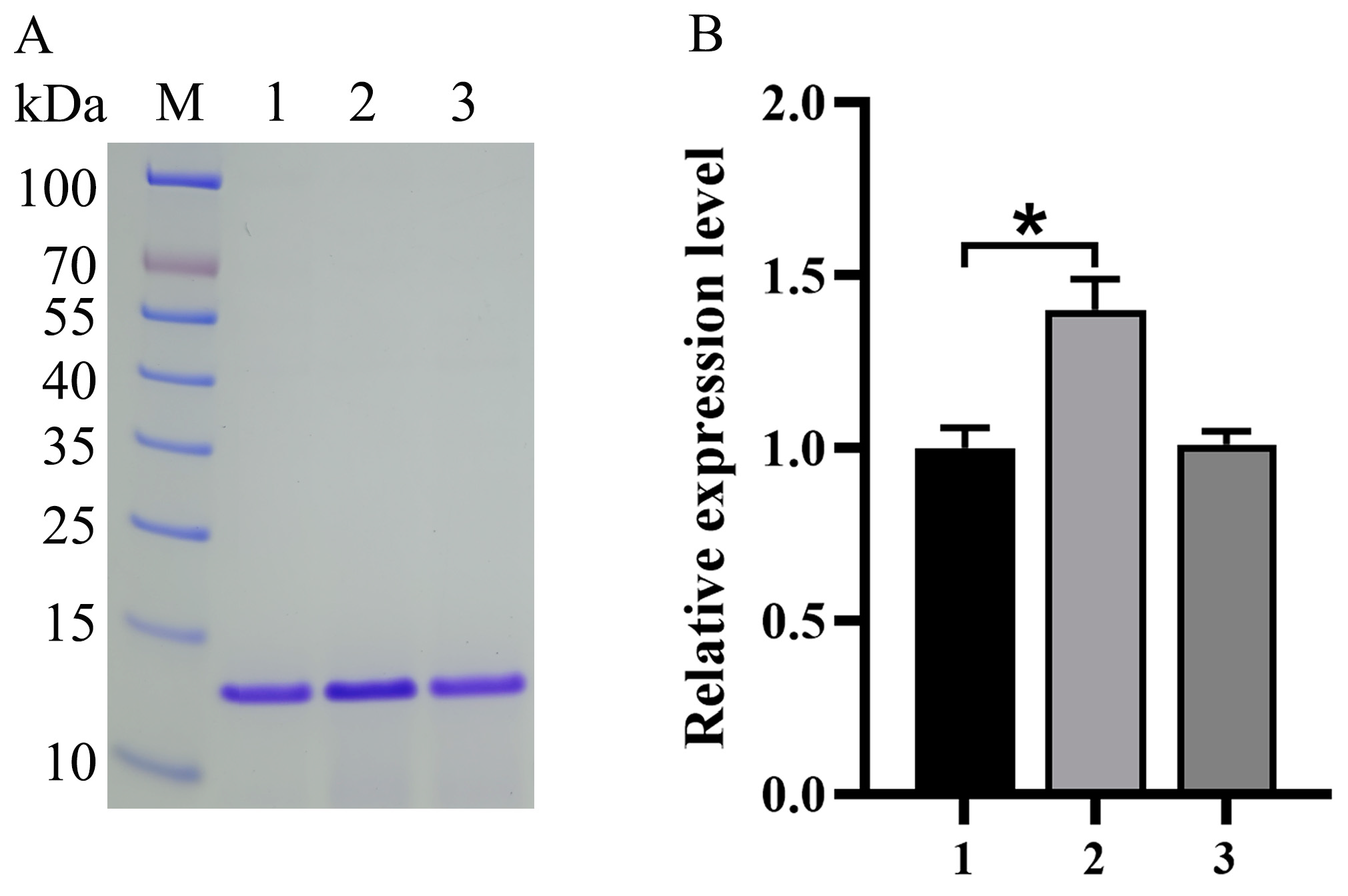
3.2. Impact of Expression Component Optimization on Va Production
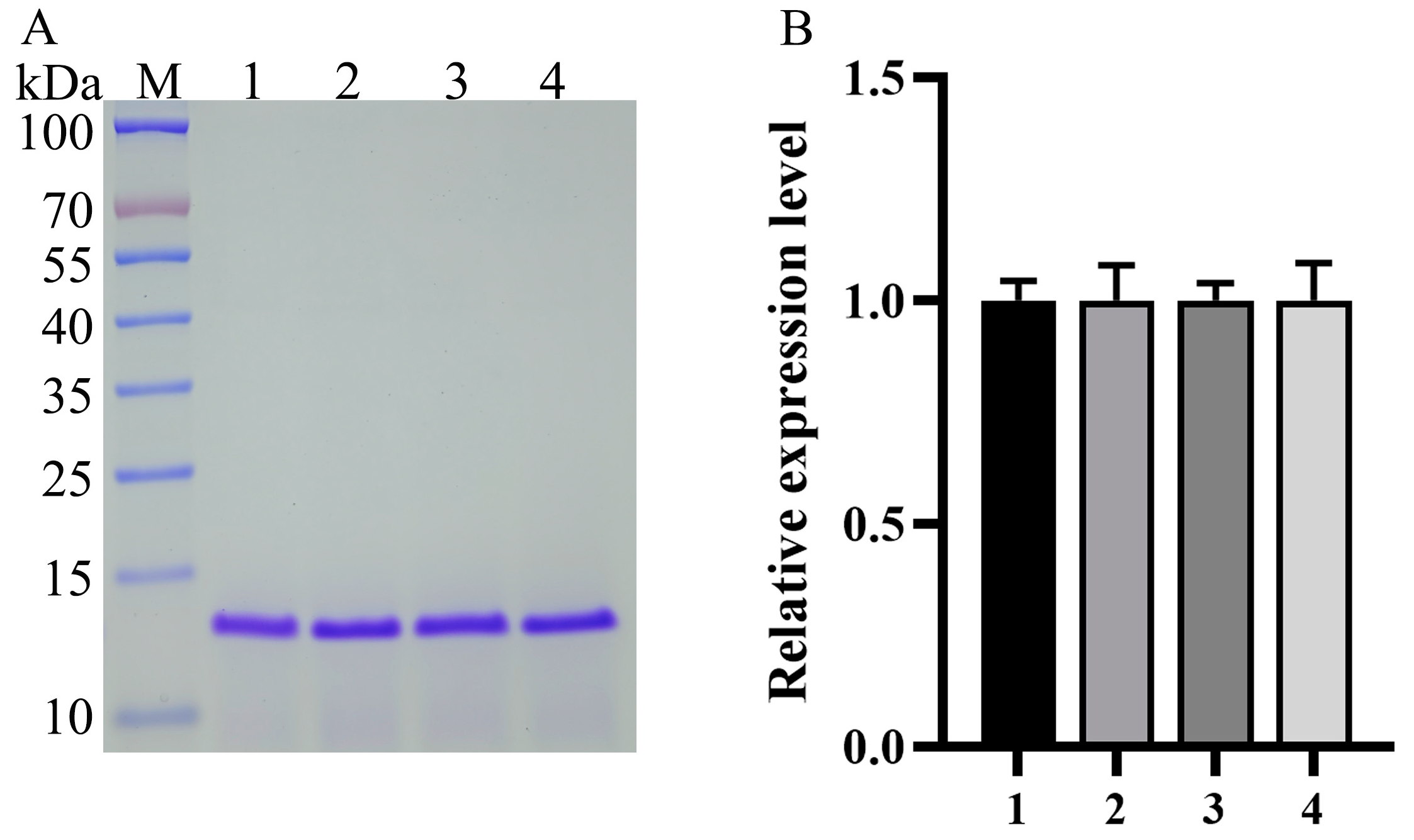
3.3. Generation and Identification of the Tandem Multicopy Expression Plasmids and Transformants
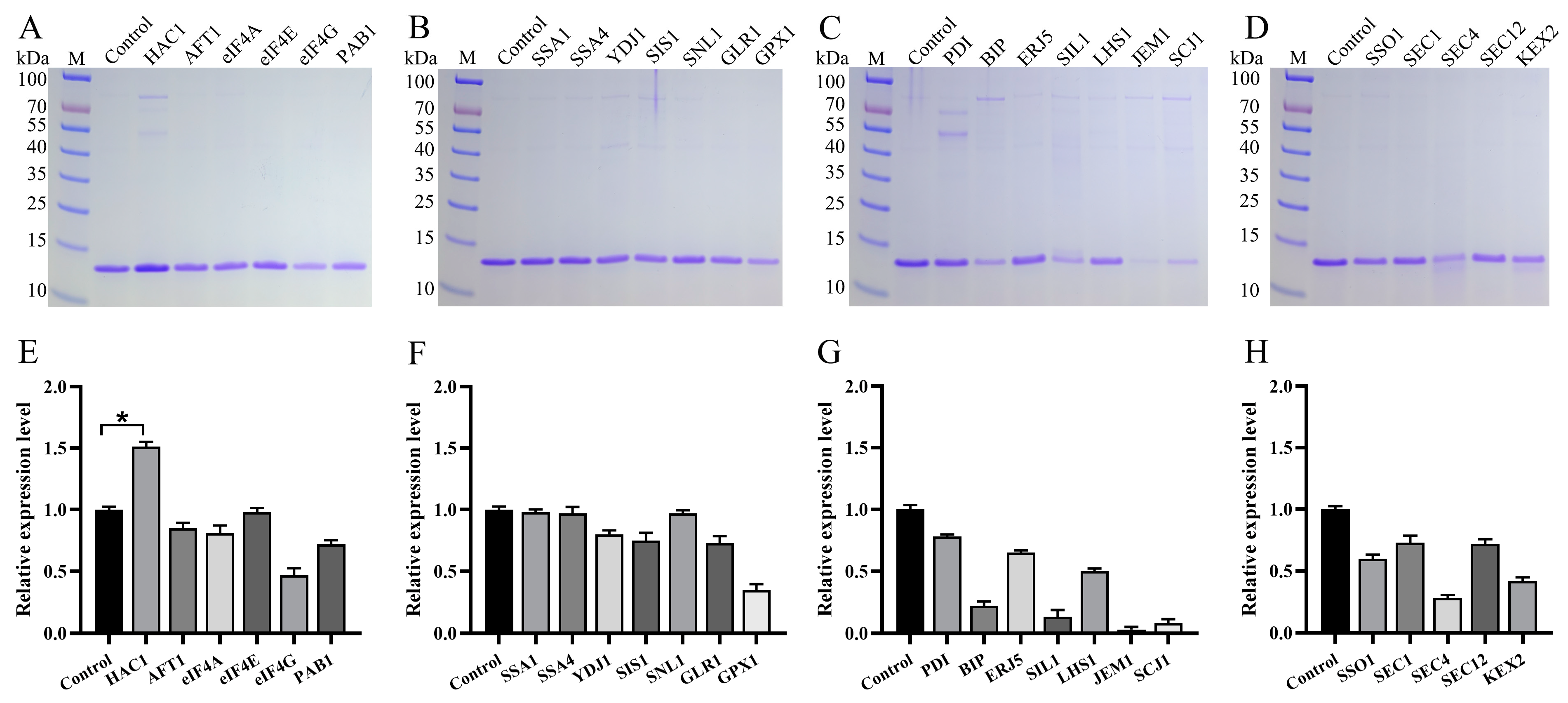
3.4. Impact of Molecular Chaperones on Va Production
3.5. Impact of Attenuated HAC1 on Va Production
3.6. High-Density Fermentation
3.7. Generation and Validation of the Packaging Strategy of High-Yielding Strain Construction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.J.; Cubitt, M.F.; Carlton, T.M.; Rodrigues-Duarte, L.; Maggiore, L.; Chai, R.; Clare, S.; Harcourt, K.; MacDonald, T.T.; Ray, K.P.; et al. Preclinical development of a bispecific TNFα/IL-23 neutralising domain antibody as a novel oral treatment for inflammatory bowel disease. Sci. Rep. 2021, 11, 19422. [Google Scholar] [CrossRef] [PubMed]
- Hanke, L.; Vidakovics Perez, L.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Karlsson Hedestam, G.B.; Hällberg, B.M. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef]
- Zhou, Z.; Vaidyanathan, G.; McDougald, D.; Kang, C.M.; Balyasnikova, I.; Devoogdt, N.; Ta, A.N.; McNaughton, B.R.; Zalutsky, M.R. Fluorine-18 labeling of the HER2-targeting single-domain antibody 2Rs15d using a residualizing label and preclinical evaluation. Mol. Imaging Biol. 2017, 19, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Wang, K.; Lu, Q.; Ji, P.; Liu, B.; Zhu, J.; Liu, Q.; Sun, Y.; Zhang, J.; Zhou, E.-M.; et al. Nanobody-horseradish peroxidase fusion protein as an ultrasensitive probe to detect antibodies against Newcastle disease virus in the immunoassay. J. Nanobiotechnol. 2019, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Shen, W.; Feng, K.; Feng, Y.; He, Z.; Li, Y.; Jiang, C.; Liu, S.; Zhu, Y.-G.; Deng, Y. Organic fertilizer potentiates the transfer of typical antibiotic resistance gene among special bacterial species. J. Hazard. Mater. 2022, 435, 128985. [Google Scholar] [CrossRef]
- Trubenová, B.; Roizman, D.; Moter, A.; Rolff, J.; Regoes, R.R. Population genetics, biofilm recalcitrance, and antibiotic resistance evolution. Trends Microbiol. 2022, 30, 841–852. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Bhattacharyya, M.; Pfannenstiel, D.M.; Nandi, A.K.; Hwang, Y.; Ho, K.; Harshey, R.M. Efflux-linked accelerated evolution of antibiotic resistance at a population edge. Mol. Cell 2022, 82, 4368–4385.e6. [Google Scholar] [CrossRef]
- Nguyen, B.-A.T.; Chen, Q.-L.; He, J.-Z.; Hu, H.-W. Microbial regulation of natural antibiotic resistance: Understanding the protist-bacteria interactions for evolution of soil resistome. Sci. Total Environ. 2020, 705, 135882. [Google Scholar] [CrossRef]
- Qin, X.; Qian, J.; Yao, G.; Zhuang, Y.; Zhang, S.; Chu, J. GAP promoter library for fine-tuning of gene expression in Pichia pastoris. Appl. Environ. Microbiol. 2011, 77, 3600–3608. [Google Scholar] [CrossRef]
- Hartner, F.S.; Ruth, C.; Langenegger, D.; Johnson, S.N.; Hyka, P.; Lin-Cereghino, G.P.; Lin-Cereghino, J.; Kovar, K.; Cregg, J.M.; Glieder, A. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 2008, 36, e76. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.S.; Yao, Q.H.; Peng, R.H.; Han, P.L.; Cheng, Z.M.; Li, Y. High level expression of a recombinant acid phytase gene in Pichia pastoris. J. Appl. Microbiol. 2005, 98, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Lin-Cereghino, G.P.; Stark, C.M.; Kim, D.; Chang, J.; Shaheen, N.; Poerwanto, H.; Agari, K.; Moua, P.; Low, L.K.; Tran, N.; et al. The effect of α-mating factor secretion signal mutations on recombinant protein expression in Pichia pastoris. Gene 2013, 519, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, H.; Yao, X.; Du, G.; Chen, J.; Kang, Z. High-yield secretory production of stable, active trypsin through engineering of the N-terminal peptide and self-degradation sites in Pichia pastoris. Bioresour. Technol. 2018, 247, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Vadhana, A.K.P.; Samuel, P.; Berin, R.M.; Krishna, J.; Kamatchi, K.; Meenakshisundaram, S. Improved secretion of Candida antarctica lipase B with its native signal peptide in Pichia pastoris. Enzyme Microb. Technol. 2013, 52, 177–183. [Google Scholar] [CrossRef]
- Liang, S.; Li, C.; Ye, Y.; Lin, Y. Endogenous signal peptides efficiently mediate the secretion of recombinant proteins in Pichia pastoris. Biotechnol. Lett. 2013, 35, 97–105. [Google Scholar] [CrossRef]
- Duan, G.; Ding, L.; Wei, D.; Zhou, H.; Chu, J.; Zhang, S.; Qian, J. Screening endogenous signal peptides and protein folding factors to promote the secretory expression of heterologous proteins in Pichia pastoris. J. Biotechnol. 2019, 306, 193–202. [Google Scholar] [CrossRef]
- Aw, R.; Polizzi, K.M. Can too many copies spoil the broth? Microb. Cell Fact. 2013, 12, 128. [Google Scholar] [CrossRef]
- Zhu, T.; Guo, M.; Tang, Z.; Zhang, M.; Zhuang, Y.; Chu, J.; Zhang, S. Efficient generation of multi-copy strains for optimizing secretory expression of porcine insulin precursor in yeast Pichia pastoris. J. Appl. Microbiol. 2009, 107, 954–963. [Google Scholar] [CrossRef]
- Delic, M.; Valli, M.; Graf, A.B.; Pfeffer, M.; Mattanovich, D.; Gasser, B. The secretory pathway: Exploring yeast diversity. FEMS Microbiol. Rev. 2013, 37, 872–914. [Google Scholar] [CrossRef]
- Bankefa, O.E.; Wang, M.; Zhu, T.; Li, Y. Hac1p homologues from higher eukaryotes can improve the secretion of heterologous proteins in the yeast Pichia pastoris. Biotechnol. Lett. 2018, 40, 1149–1156. [Google Scholar] [CrossRef]
- Guerfal, M.; Ryckaert, S.; Jacobs, P.P.; Ameloot, P.; Van Craenenbroeck, K.; Derycke, R.; Callewaert, N. The HAC1 gene from Pichia pastoris: Characterization and effect of its overexpression on the production of secreted, surface displayed and membrane proteins. Microb. Cell Fact. 2010, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in Pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Goeckeler, J.; Schekman, R. BiP and Sec63p are required for both co-and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1995, 92, 9643–9646. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, J.; Chen, J.; Zhang, D.; Zhang, Y.; Qiao, X.; Yu, X.; Zheng, Q.; Hou, J. Optimized expression of classical swine fever virus E2 protein via combined strategy in Pichia pastoris. Protein Expr. Purif. 2020, 167, 105527. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.; Xi, D.; Zhang, J.; Wang, X.; Mao, R.; Zhang, Y.; Wang, J. Multiple copies of the target gene enhances plectasin secretion in Pichia pastoris X-33. Process Biochem. 2015, 50, 553–560. [Google Scholar] [CrossRef]
- Li, D.; Zhang, B.; Li, S.; Zhou, J.; Cao, H.; Huang, Y.; Cui, Z. A novel vector for construction of markerless multicopy overexpression transformants in Pichia pastoris. Front. Microbiol. 2017, 8, 1698. [Google Scholar] [CrossRef]
- Ulrichts, H.; Silence, K.; Schoolmeester, A.; de Jaegere, P.; Rossenu, S.; Roodt, J.; Priem, S.; Lauwereys, M.; Casteels, P.; Van Bockstaele, F.; et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood J. Am. Soc. Hematol. 2011, 118, 757–765. [Google Scholar] [CrossRef]
- Coppieters, K.; Dreier, T.; Silence, K.; Haard, H.D.; Lauwereys, M.; Casteels, P.; Beirnaert, E.; Jonckheere, H.; Wiele, C.V.D.; Staelens, L.; et al. Formatted anti-tumor necrosis factor α VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis. Rheum. 2006, 54, 1856–1866. [Google Scholar] [CrossRef]
- Bobkov, V.; Zarca, A.M.; Van Hout, A.; Arimont, M.; Doijen, J.; Bialkowska, M.; Toffoli, E.; Klarenbeek, A.; van der Woning, B.; van der Vliet, H.J.; et al. Nanobody-Fc constructs targeting chemokine receptor CXCR4 potently inhibit signaling and CXCR4-mediated HIV-entry and induce antibody effector functions. Biochem. Pharmacol. 2018, 158, 413–424. [Google Scholar] [CrossRef]
- Jiang, H.; Ni, H.; Zhang, P.; Guo, X.; Wu, M.; Shen, H.; Wang, J.; Wu, W.; Wu, Z.; Ding, J.; et al. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology 2021, 10, 1943180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ni, H.; Zhou, S.; He, K.; Gao, Y.; Wu, W.; Wu, M.; Wu, Z.; Qiu, X.; Zhou, Y. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 2021, 70, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, M.; Ververken, C.; Beirnaert, E.; Hoefman, S.; Kolkman, J.; Vierboom, M.; Breedveld, E.; ‘t Hart, B.; Poelmans, S.; Bontinck, L.; et al. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob. Agents Chemother. 2016, 60, 6–13. [Google Scholar] [CrossRef]
- Pan, H.; Su, Y.; Xie, Y.; Wang, W.; Qiu, W.; Chen, W.; Lu, W.; Lu, Z.; Wang, W.; Shang, A. Everestmab, a novel long-acting GLP-1/anti GLP-1R nanobody fusion protein, exerts potent anti-diabetic effects. Artif. Cells Nanomed. Biotechnol. 2020, 48, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.; Ma, L.; Li, G.; Zhu, M.; Qiao, P.; Li, X.; Zhang, H.; Zhang, Y.; Chen, Y.; Ji, W. A potent neutralizing nanobody against SARS-CoV-2 with inhaled delivery potential. MedComm 2021, 2, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Song, L.; Peng, Q.; Zhu, Q.; Shi, X.; Xu, M.; Wang, Q.; Zhang, Y.; Cai, M. A programmable high-expression yeast platform responsive to user-defined signals. Sci. Adv. 2022, 8, eabl5166. [Google Scholar] [CrossRef]
- Ruth, C.; Buchetics, M.; Vidimce, V.; Kotz, D.; Naschberger, S.; Mattanovich, D.; Pichler, H.; Gasser, B. Pichia pastoris Aft1-a novel transcription factor, enhancing recombinant protein secretion. Microb. Cell Fact. 2014, 13, 120. [Google Scholar] [CrossRef]
- Huang, H.; Liang, Q.; Wang, Y.; Chen, J.; Kang, Z. High-level constitutive expression of leech hyaluronidase with combined strategies in recombinant Pichia pastoris. Appl. Microbiol. Biotechnol. 2020, 104, 1621–1632. [Google Scholar] [CrossRef]
- Staudacher, J.; Rebnegger, C.; Dohnal, T.; Landes, N.; Mattanovich, D.; Gasser, B. Going beyond the limit: Increasing global translation activity leads to increased productivity of recombinant secreted proteins in Pichia pastoris. Metab. Eng. 2022, 70, 181–195. [Google Scholar] [CrossRef]
- Zahrl, R.J.; Prielhofer, R.; Ata, Ö.; Baumann, K.; Mattanovich, D.; Gasser, B. Pushing and pulling proteins into the yeast secretory pathway enhances recombinant protein secretion. Metab. Eng. 2022, 74, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Ben Azoun, S.; Belhaj, A.E.; Göngrich, R.; Gasser, B.; Kallel, H. Molecular optimization of rabies virus glycoprotein expression in Pichia pastoris. Microb. Biotechnol. 2016, 9, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Chen, F.; Su, S.; Duan, Z.; Chen, Y.; Li, H.; Jin, J. Effects of co-overexpression of secretion helper factors on the secretion of a HSA fusion protein (IL2-HSA) in Pichia pastoris. Yeast 2016, 33, 587–600. [Google Scholar] [CrossRef]
- Liu, S.H.; Chou, W.I.; Sheu, C.C.; Chang, M.D. Improved secretory production of glucoamylase in Pichia pastoris by combination of genetic manipulations. Biochem. Biophys. Res. Commun. 2005, 326, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kuang, Y.; Li, H.; Liu, Y.; Hui, X.; Li, P.; Jiang, Z.; Zhou, Y.; Wang, Y.; Xu, A.; et al. Enhanced production of recombinant secretory proteins in Pichia pastoris by optimizing Kex2 P1’site. PLoS ONE 2013, 8, e75347. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, J.C.; Koskela, E.V.; Frey, A.D. Enhancing antibody folding and secretion by tailoring the Saccharomyces cerevisiae endoplasmic reticulum. Microb. Cell Fact. 2016, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, Y.; Zheng, X.; Pang, N.; Liao, X.; Liu, X.; Huang, Y.; Liang, S. Combined strategies for improving expression of Citrobacter amalonaticus phytase in Pichia pastoris. BMC Biotechnol. 2015, 15, 88. [Google Scholar] [CrossRef]
- Yu, S.; Miao, L.; Huang, H.; Li, Y.; Zhu, T. High-level production of glucose oxidase in Pichia pastoris: Effects of Hac1p overexpression on cell physiology and enzyme expression. Enzyme Microb. Technol. 2020, 141, 109671. [Google Scholar] [CrossRef]
- Rakestraw, J.A.; Sazinsky, S.L.; Piatesi, A.; Antipov, E.; Wittrup, K.D. Directed evolution of a secretory leader for the improved expression of heterologous proteins and full-length antibodies in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2009, 103, 1192–1201. [Google Scholar] [CrossRef]
- Clare, J.J.; Romanes, M.A.; Rayment, F.B.; Rowedder, J.E.; Smith, M.A.; Payne, M.M.; Sreekrishna, K.; Henwood, C.A. Production of mouse epidermal growth factor in yeast: High-level secretion using Pichia pastoris strains containing multiple gene copies. Gene 1991, 105, 205–212. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Abbreviated Names of the PAOX1 Mutants | Promoter Strength | Signal Peptide |
|---|---|---|---|
| pLD-AOXα | AOX1 (WT) | 100% (WT) | α-factor |
| pLD-P1α | 133p | 133% | α-factor |
| pLD-P2α | 160p | 160% | α-factor |
| pLD-P2S1 | 160p | 160% | MF4I |
| pLD-P2S2 | 160p | 160% | α-factor Δ57–70 |
| pLD-P2S3 | 160p | 160% | α-factor Δ57–70 plus HL28 |
| pLD-P2S4 | 160p | 160% | nSB |
| pLD-P2S5 | 160p | 160% | Dse4 |
| pLD-P2S6 | 160p | 160% | Msb2 |
| pLD-P2S7 | 160p | 160% | Gas1 |
| pLD-AOX | AOX1 (WT) | 100% (WT) | None |
| pLD-6p | 6p | 6% | None |
| pLD-14p | 14p | 14% | None |
| pLD-30p | 30p | 30% | None |
| pLD-55p | 55p | 55% | None |
| pLD-75p | 75p | 75% | None |
| pLD-160p | 160p | 160% | None |
| Name | Function | Major Subcellular Localization | Genomic Number |
|---|---|---|---|
| HAC1 | Transcription factor | Nucleus | PAS_chr1-1_0381 |
| AFT1 | Transcription factor | Nucleus | PAS_chr1-4_0361 |
| eIF4A | Eukaryotic translation initiation factor | Cytoplasm | PAS_chr3_0595 |
| eIF4E | Eukaryotic translation initiation factor | Cytoplasm | PAS_chr3_0972 |
| eIF4G | Eukaryotic translation initiation factor | Cytoplasm | PAS_chr1-1_0053 |
| PAB1 | Poly(A) binding protein | Cytoplasm | PAS_chr2-1_0097 |
| SSA1 | Hsp70 | Cytoplasm | PAS_chr4_0552 |
| SSA4 | Hsp70 | Cytoplasm | PAS_chr3_0230 |
| YDJ1 | Hsp40 | Cytoplasm | PAS_chr2-2_0066 |
| SIS1 | Hsp40 | Cytoplasm | PAS_chr2-2_0151 |
| SNL1 | Nucleotide exchange factor | Cytoplasm | PAS_chr1-4_0091 |
| GPX1 | Glutathione peroxidase | Cytoplasm | PAS_chr2-2_0382 |
| GLR1 | Glutathione reductase | Mitochondrion | PAS_chr3_1011 |
| PDI | Disulfide isomerase | ER | PAS_chr4_0844 |
| BIP | Hsp70 | ER | PAS_chr2-1_0140 |
| SIL1 | Nucleotide exchange factor | ER | PAS_chr1-1_0237 |
| LHS1 | Nucleotide exchange factor | ER | PAS_chr1-3_0063 |
| ERJ5 | Hsp40 | ER | PAS_chr1-3_0134 |
| JEM1 | Hsp40 | ER | PAS_chr2-2_0015 |
| SCJ1 | Hsp40 | ER | PAS_chr1-3_0174 |
| SSO1 | Plasma membrane t-SNARE protein | Golgi | PAS_chr1-4_0294 |
| SEC1 | Plasma membrane t-SNARE protein | Golgi | PAS_chr4_0134 |
| SEC4 | Rab family of GTP binding protein | Golgi | PAS_chr3_0143 |
| SEC12 | Nucleotide exchange factor | Golgi | PAS_chr4_0606 |
| KEX2 | Endoprotease | Golgi | PAS_chr2-1_0304 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Li, B.; Zhao, S.; Liu, J.; Li, D. A Universal Strategy for the Efficient Expression of Nanobodies in Pichia pastoris. Fermentation 2024, 10, 37. https://doi.org/10.3390/fermentation10010037
Zheng Y, Li B, Zhao S, Liu J, Li D. A Universal Strategy for the Efficient Expression of Nanobodies in Pichia pastoris. Fermentation. 2024; 10(1):37. https://doi.org/10.3390/fermentation10010037
Chicago/Turabian StyleZheng, Yiheng, Bingkun Li, Shida Zhao, Jiawei Liu, and Ding Li. 2024. "A Universal Strategy for the Efficient Expression of Nanobodies in Pichia pastoris" Fermentation 10, no. 1: 37. https://doi.org/10.3390/fermentation10010037
APA StyleZheng, Y., Li, B., Zhao, S., Liu, J., & Li, D. (2024). A Universal Strategy for the Efficient Expression of Nanobodies in Pichia pastoris. Fermentation, 10(1), 37. https://doi.org/10.3390/fermentation10010037