
A New Concept for the Rapid Development of Digital Twin Core Models for Bioprocesses in Various Reactor Designs


Abstract
1. Introduction
“Digital twins are (…) digital replications of living as well as non-living entities that enable data to be seamlessly transmitted between the physical and virtual worlds”.
2. Materials and Methods
3. Results
- Adding, exchanging or removing submodels requires changes throughout the entire source code of the DTs core model. A manual adaptation of the model structure takes at least a few hours, or even multiple days, of work for more elaborate changes;
- Non-ideal flow patterns in bioreactors may have an impact on the kinetics, performance and dynamics of the process under consideration. Thus, for a realistic representation of these effects in DTs, possibilities should be created to represent non-ideal reactor behaviour and/or different reactor types with the reactor submodel;
- The numerical solution of large mechanistic models, consisting of systems of a high number of nonlinear coupled differential equations, requires high computational effort. It is particularly important to keep the computation times of DTs, especially for their parameterisation and application in process optimisation, as short as possible.
3.1. Characteristics of the New Software Tool Concept for Automated Bioprocess DT Core Model Development
3.1.1. Biokinetic Submodel
3.1.2. Physico-Chemical Submodel
3.1.3. Reactor Submodel
- Only the required models are implemented into the final DT core model. The user can decide which models to include. Temperature, DO and pH can be defined as fixed values or fixed profiles if a calculation is not necessary;
- Only necessary double-sigmoidal functions are implemented into the DT. These functions demand a high computational effort since two exponential functions must be solved (Equation (3)). For this purpose, a user-predefined configuration file comprising the parameters of the double-sigmoidal functions is scanned using the software tool concept. If the parameters are defined in such a way that the function yields the neutral element for multiplications (yh = ymid = yl = 1), the function is not transferred into the DT core model because the result of the function equals one in any case;
- A fast calculation mode is selectable by the user. The temperature submodel (based on dynamic energy and mass balances) and the DO submodel (based on dynamic mass balances and mass transfer theory, see reference [31]) have faster time constants (in their differential equations) compared to the biokinetic submodel and are thus decisive for the number of necessary calculation steps. The fast calculation mode enables a more than 80% shorter calculation time at the expense of simulation accuracy by reducing necessary calculation steps. In the temperature model, the fast calculation mode lowers heat transfer coefficients and thus slows down the heat transfer rates. In the DO model, for the fast calculation mode, the differential equations for the calculation of the DO and the gas phase composition are replaced by algebraic equations.
3.2. Application of the New Software Tool Concept for the Development of Bioprocess DTs
3.2.1. DT Core Model for the Cultivation of S. cerevisiae in Two Coupled Parallel 1 L STRs
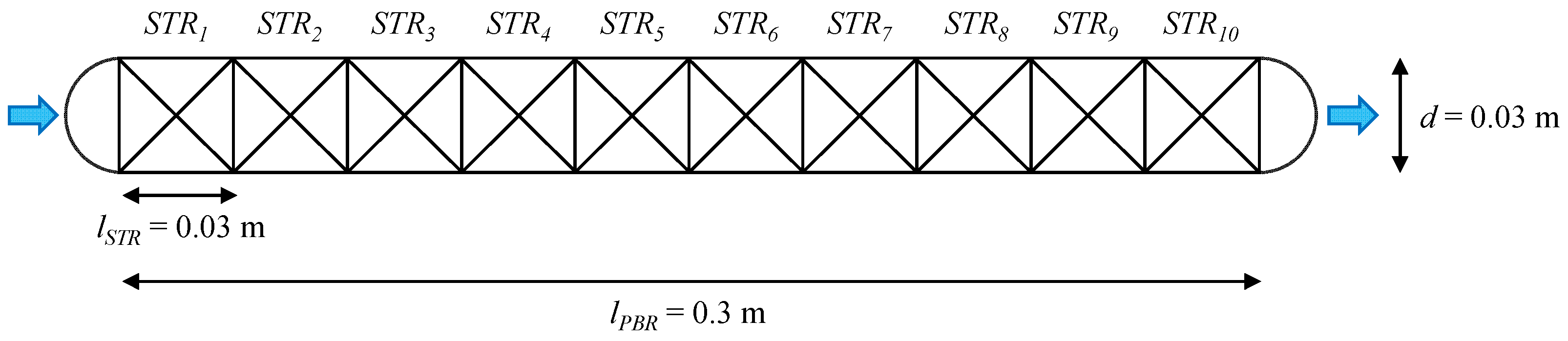
3.2.2. DT Core Model for Enzymatic Hydrolysis Processes in a PBR
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Appl, C.; Moser, A.; Baganz, F.; Hass, V.C. Digital Twins for Bioprocess Control Strategy Development and Realisation. Adv. Biochem. Eng. Biotechnol. 2020, 177, 63–94. [Google Scholar] [CrossRef]
- Grieves, M. Origins of the Digital Twin Concept; Working Paper; Florida Institute of Technology: Melbourne, FL, USA, 2016. [Google Scholar]
- Glaessgen, E.; Stargel, D. The Digital Twin Paradigm for Future NASA and U.S. Air Force Vehicles. In Proceedings of the 53rd AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics and Materials Conference, Honolulu, HI, USA, 23–26 April 2012. [Google Scholar] [CrossRef]
- El Saddik, A. Digital Twins: The Convergence of Multimedia Technologies. IEEE MultiMedia 2018, 25, 87–92. [Google Scholar] [CrossRef]
- He, R.; Chen, G.; Dong, C.; Sun, S.; Shen, X. Data-driven digital twin technology for optimized control in process systems. ISA Trans. 2019, 95, 221–234. [Google Scholar] [CrossRef]
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef]
- Blesgen, A.; Hass, V.C. Operator Training Simulator for Anaerobic Digestion Processes. IFAC Proc. Vol. 2010, 43, 353–358. [Google Scholar] [CrossRef]
- Moser, A.; Appl, C.; Brüning, S.; Hass, V.C. Mechanistic Mathematical Models as a Basis for Digital Twins. Adv. Biochem. Eng. Biotechnol. 2020, 176, 133–180. [Google Scholar] [CrossRef]
- Ingenieurbüro Dr.-Ing.Schoop GmbH. WinErs; Ingenieurbüro Dr.-Ing.Schoop GmbH: Hamburg, Germany, 2018. [Google Scholar]
- Nagy, Z.K. Model based control of a yeast fermentation bioreactor using optimally designed artificial neural networks. Chem. Eng. J. 2007, 127, 95–109. [Google Scholar] [CrossRef]
- Chen, L.; Nguang, S.K.; Chen, X.D.; Li, X.M. Modelling and optimization of fed-batch fermentation processes using dynamic neural networks and genetic algorithms. Biochem. Eng. J. 2004, 22, 51–61. [Google Scholar] [CrossRef]
- Grahovac, J.; Jokić, A.; Dodić, J.; Vučurović, D.; Dodić, S. Modelling and prediction of bioethanol production from intermediates and byproduct of sugar beet processing using neural networks. Renew. Energy 2016, 85, 953–958. [Google Scholar] [CrossRef]
- Konstantinov, K.B.; Yoshida, T. An expert approach for control of fermentation processes as variable structure plants. J. Ferment. Bioeng. 1990, 70, 48–57. [Google Scholar] [CrossRef]
- Sahakyan, M.; Aung, Z.; Rahwan, T. Explainable Artificial Intelligence for Tabular Data: A Survey. IEEE Access 2021, 9, 135392–135422. [Google Scholar] [CrossRef]
- Treloar, N.J.; Fedorec, A.J.H.; Ingalls, B.; Barnes, C.P. Deep reinforcement learning for the control of microbial co-cultures in bioreactors. PLoS Comput. Biol. 2020, 16, e1007783. [Google Scholar] [CrossRef] [PubMed]
- Von Stosch, M.; Davy, S.; Francois, K.; Galvanauskas, V.; Hamelink, J.-M.; Luebbert, A.; Mayer, M.; Oliveira, R.; O’Kennedy, R.; Rice, P.; et al. Hybrid modeling for quality by design and PAT-benefits and challenges of applications in biopharmaceutical industry. Biotechnol. J. 2014, 9, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, L.A. Fuzzy sets. Inf. Control 1965, 8, 338–353. [Google Scholar] [CrossRef]
- Ying, H. Essentials of fuzzy modeling and control. J. Am. Soc. Inf. Sci. 1995, 46, 791–792. [Google Scholar] [CrossRef]
- Karakuzu, C.; Türker, M.; Öztürk, S. Modelling, on-line state estimation and fuzzy control of production scale fed-batch baker’s yeast fermentation. Control. Eng. Pract. 2006, 14, 959–974. [Google Scholar] [CrossRef]
- Horiuchi, J.-I. Fuzzy modeling and control of biological processes. J. Biosci. Bioeng. 2002, 94, 574–578. [Google Scholar] [CrossRef]
- Monod, J. The Growth of Bacterial Cultures. Annu. Rev. Microbiol. 1949, 3, 371–394. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M.L. Kinetik der Invertinwirkung. Biochem. Ztg. 1913, 49, 333–369. [Google Scholar]
- Cornish-Bowden, A. One hundred years of Michaelis–Menten kinetics. Perspect. Sci. 2015, 4, 3–9. [Google Scholar] [CrossRef]
- González-Figueredo, C.; Flores-Estrella, R.A.; Rojas-Rejón, O.A. Fermentation: Metabolism, Kinetic Models, and Bioprocessing. In Current Topics in Biochemical Engineering; Shiomi, N., Ed.; IntechOpen: London, UK, 2019; ISBN 978-1-83881-209-6. [Google Scholar]
- Craven, S.; Shirsat, N.; Whelan, J.; Glennon, B. Process model comparison and transferability across bioreactor scales and modes of operation for a mammalian cell bioprocess. Biotechnol. Prog. 2013, 29, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, B.J.; Taylor, C.N.; Ushio, M.; Leigh, J.R.; Kalganova, T.; Baganz, F. Intelligent modelling of bioprocesses: A comparison of structured and unstructured approaches. Bioprocess Biosyst. Eng. 2004, 26, 353–359. [Google Scholar] [CrossRef]
- Esener, A.A.; Roels, J.A.; Kossen, N.W. Theory and applications of unstructured growth models: Kinetic and energetic aspects. Biotechnol. Bioeng. 1983, 25, 2803–2841. [Google Scholar] [CrossRef] [PubMed]
- Shuler, M.L.; Leung, S.; Dick, C.C. A Mathematical Model for the Growth of a Single Bacterial Cell. Ann. N. Y. Acad. Sci. 1979, 326, 35–52. [Google Scholar] [CrossRef]
- Nielsen, J.; Nikolajsen, K.; Villadsen, J. Structured modeling of a microbial system: I. A theoretical study of lactic acid fermentation. Biotechnol. Bioeng. 1991, 38, 1–10. [Google Scholar] [CrossRef]
- Appl, C.; Baganz, F.; Hass, V.C. Development of a Digital Twin for Enzymatic Hydrolysis Processes. Processes 2021, 9, 1734. [Google Scholar] [CrossRef]
- Brüning, S. Development of a Generalized Process Model for Optimization of Biotechnological Processes. Ph.D. Thesis, Jacobs University, Bremen, Germany, 2016. [Google Scholar]
- Hass, V.C.; Kuhnen, F.; Schoop, K.-M. An environment for the development of operator training systems (OTS) from chemical engineering models. Comput. Aided Chem. Eng. 2005, 20, 289–293. [Google Scholar] [CrossRef]
- R Core Team. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2014. [Google Scholar]
- Hirschmann, R. Evaluating the Potential of Anaerobic Production of Ethyl(3)Hydroxybutyrate for Integration in Biorefineries. Ph.D. Thesis, University College London, London, UK, 2022. [Google Scholar]
- Gerlach, I.; Brüning, S.; Gustavsson, R.; Mandenius, C.-F.; Hass, V.C. Operator training in recombinant protein production using a structured simulator model. J. Biotechnol. 2014, 177, 53–59. [Google Scholar] [CrossRef]
- Kuntzsch, S. Energy Efficiency Investigations with a New Operator Training Simulator for Biorefineries. Ph.D. Thesis, Jacobs University, Bremen, Germany, 2014. [Google Scholar]
- Laurí, D.; Lennox, B.; Camacho, J. Model predictive control for batch processes: Ensuring validity of predictions. J. Process. Control. 2014, 24, 239–249. [Google Scholar] [CrossRef]
- Mears, L.; Stocks, S.M.; Sin, G.; Gernaey, K.V. A review of control strategies for manipulating the feed rate in fed-batch fermentation processes. J. Biotechnol. 2017, 245, 34–46. [Google Scholar] [CrossRef]
- Moser, A.; Kuchemüller, K.B.; Deppe, S.; Hernández Rodríguez, T.; Frahm, B.; Pörtner, R.; Hass, V.C.; Möller, J. Model-assisted DoE software: Optimization of growth and biocatalysis in Saccharomyces cerevisiae bioprocesses. Bioprocess Biosyst. Eng. 2021, 44, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Mandenius, C.-F.; Brundin, A. Bioprocess optimization using design-of-experiments methodology. Biotechnol. Prog. 2008, 24, 1191–1203. [Google Scholar] [CrossRef]
- Candioti, L.V.; de Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental design and multiple response optimization. Using the desirability function in analytical methods development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Kuchemüller, K.B.; Steinmetz, T.; Koopmann, K.S.; Pörtner, R. Model-assisted Design of Experiments as a concept for knowledge-based bioprocess development. Bioprocess Biosyst. Eng. 2019, 42, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, P.; Junne, S. Scale-down simulators for metabolic analysis of large-scale bioprocesses. Curr. Opin. Biotechnol. 2010, 21, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, N.M.; Kossen, N.W. Dissolved oxygen concentration profiles in a production-scale bioreactor. Biotechnol. Bioeng. 1984, 26, 546–550. [Google Scholar] [CrossRef]
- Hewitt, C.J.; Nienow, A.W. The scale-up of microbial batch and fed-batch fermentation processes. Adv. Appl. Microbiol. 2007, 62, 105–135. [Google Scholar] [CrossRef]
- Enfors, S.O.; Jahic, M.; Rozkov, A.; Xu, B.; Hecker, M.; Jürgen, B.; Krüger, E.; Schweder, T.; Hamer, G.; O’Beirne, D.; et al. Physiological responses to mixing in large scale bioreactors. J. Biotechnol. 2001, 85, 175–185. [Google Scholar] [CrossRef]
- George, S.; Larsson, G.; Olsson, K.; Enfors, S.-O. Comparison of the Baker’s yeast process performance in laboratory and production scale. Bioprocess Eng. 1998, 18, 135–142. [Google Scholar] [CrossRef]
- Larsson, G.; Trnkvist, M.; Wernersson, E.S.; Trgrdh, C.; Noorman, H.; Enfors, S.-O. Substrate gradients in bioreactors: Origin and consequences. Bioprocess Eng. 1996, 14, 281–289. [Google Scholar] [CrossRef]
- Formenti, L.R.; Nørregaard, A.; Bolic, A.; Hernandez, D.Q.; Hagemann, T.; Heins, A.-L.; Larsson, H.; Mears, L.; Mauricio-Iglesias, M.; Krühne, U.; et al. Challenges in industrial fermentation technology research. Biotechnol. J. 2014, 9, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Sweere, A.P.J.; Matla, Y.A.; Zandvliet, J.; Luyben, K.C.A.M.; Kossen, N.W.F. Experimental simulation of glucose fluctuations. Appl. Microbiol. Biotechnol. 1988, 28, 109–115. [Google Scholar] [CrossRef]
- Sandoval-Basurto, E.A.; Gosset, G.; Bolívar, F.; Ramírez, O.T. Culture of Escherichia coli under dissolved oxygen gradients simulated in a two-compartment scale-down system: Metabolic response and production of recombinant protein. Biotechnol. Bioeng. 2005, 89, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lara, A.R.; Galindo, E.; Ramírez, O.T.; Palomares, L.A. Living with heterogeneities in bioreactors: Understanding the effects of environmental gradients on cells. Mol. Biotechnol. 2006, 34, 355–381. [Google Scholar] [CrossRef]
- Fiechter, A.; Fuhrmann, G.F.; Käppeli, O. Regulation of Glucose Metabolism in Growing Yeast Cells. Adv. Microb. Physiol. 1981, 22, 123–183. [Google Scholar] [CrossRef]
- Sonnleitner, B.; Käppeli, O. Growth of Saccharomyces cerevisiae is controlled by its limited respiratory capacity: Formulation and verification of a hypothesis. Biotechnol. Bioeng. 1986, 28, 927–937. [Google Scholar] [CrossRef]
- Bai, F.W.; Anderson, W.A.; Moo-Young, M. Ethanol fermentation technologies from sugar and starch feedstocks. Biotechnol. Adv. 2008, 26, 89–105. [Google Scholar] [CrossRef]
- Larsson, C.; von Stockar, U.; Marison, I.; Gustafsson, L. Growth and Metabolism of Saccharomyces cerevisiae in Chemostat Cultures under Carbon-, Nitrogen-, or Carbon- and Nitrogen-Limiting Conditions. J. Bacteriol. 1993, 175, 4809–4816. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Nienow, A.W. Scale-Up, Stirred Tank Reactors. In Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 1–38. [Google Scholar] [CrossRef]
- Van’t Riet, K.; Tramper, J. Basic Bioreactor Design, 1st ed.; CRC Press: Boca Raton, FL, USA, 1991; ISBN 9781482293333. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Submodel | Model | Options/Specification |
|---|---|---|
| Biokinetic | Microbiological | Saccharomyces cerevisiae |
| Cyathus striatus | ||
| Lactobacillus delbrueckii | ||
| Escherichia coli | ||
| Mammalian cells | Hybridoma cells | |
| Chinese hamster ovary (CHO) cells | ||
| Biocatalysis | Whole-cell biocatalysis | |
| Enzymatic reactions | Starch hydrolysis | |
| Proteolysis | ||
| Physico-chemical | Gas-phase | Calculation |
| pH value | Calculation | |
| Fixed profile | ||
| Fixed value | ||
| Dissolved oxygen | Algebraic equations | |
| Differential equations | ||
| Fixed profile | ||
| Fixed value | ||
| Temperature | Calculation | |
| Fixed profile | ||
| Fixed value | ||
| Foam level | Calculation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moser, A.; Appl, C.; Pörtner, R.; Baganz, F.; Hass, V.C. A New Concept for the Rapid Development of Digital Twin Core Models for Bioprocesses in Various Reactor Designs. Fermentation 2024, 10, 463. https://doi.org/10.3390/fermentation10090463
Moser A, Appl C, Pörtner R, Baganz F, Hass VC. A New Concept for the Rapid Development of Digital Twin Core Models for Bioprocesses in Various Reactor Designs. Fermentation. 2024; 10(9):463. https://doi.org/10.3390/fermentation10090463
Chicago/Turabian StyleMoser, André, Christian Appl, Ralf Pörtner, Frank Baganz, and Volker C. Hass. 2024. "A New Concept for the Rapid Development of Digital Twin Core Models for Bioprocesses in Various Reactor Designs" Fermentation 10, no. 9: 463. https://doi.org/10.3390/fermentation10090463
APA StyleMoser, A., Appl, C., Pörtner, R., Baganz, F., & Hass, V. C. (2024). A New Concept for the Rapid Development of Digital Twin Core Models for Bioprocesses in Various Reactor Designs. Fermentation, 10(9), 463. https://doi.org/10.3390/fermentation10090463