
Improving the Synthesis Efficiency of Amino Acids Such as L-Lysine by Assembling Artificial Cellulosome Elements Dockerin Protein In Vivo

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Selection of Key Components of Cellulosomes
2.3. Expression and Purification of DocA and Coh
2.4. Mutation and Verification of DocA
2.5. Detection of DocA and Coh Binding
2.6. BiFC-FC
2.7. MD Simulation
2.8. Assembly and Detection of Key Enzymes in L-Lysine Fermentation
2.9. Assembly and Detection of Key Enzymes in β-Alanine Fermentation
3. Results and Discussion
3.1. Selection of The Self-Assembly System
3.2. Selection and Expression of Key Components of Cellulosomes
3.3. Mutation of Coh and DocA
3.4. In Vitro Verification of DocA Mutant Binding to Coh
3.5. In Vivo Verification of DocA Mutant Binding to Coh
3.6. MD Simulation and Structural Analysis of DocA Mutants
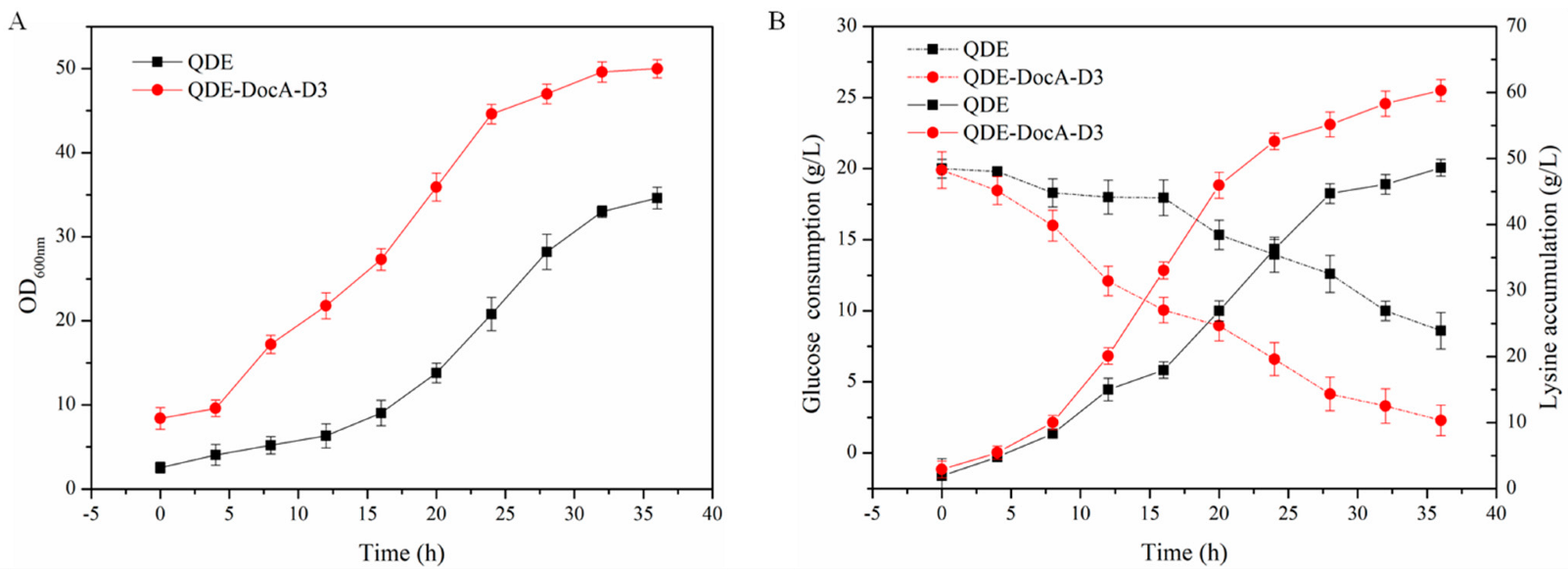
3.7. Efficient Assembly and Detection of Key Enzymes for L-Lysine Production
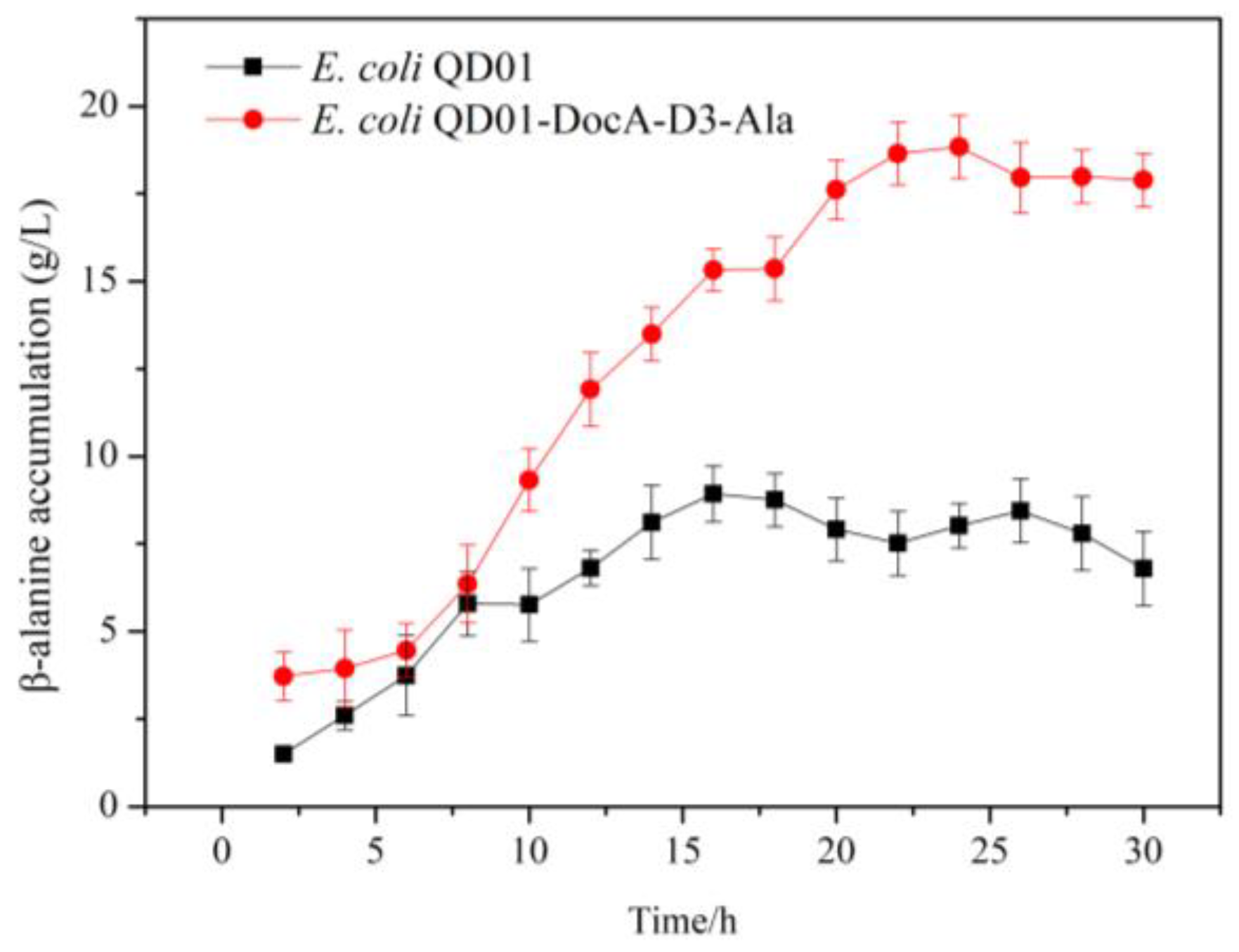
3.8. Efficient Assembly and Detection of Key Enzymes for β-Alanine Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, C.K.; Pedley, A.M.; He, J.X.; Johnson, J.L.; Yaron, T.M.; Cantley, L.C.; Benkovic, S.J. Biocatalytic cascades operating on macromolecular scaffolds and in confined environments. Nat. Catal. 2020, 3, 256–273. [Google Scholar]
- Fletcher, J.M.; Harniman, R.L.; Barnes, F.R.H.; Boyle, A.L.; Collins, A.; Mantell, J.; Sharp, T.H.; Antognozzi, M.; Booth, P.J.; Linden, N.; et al. Self-assembling cages from coiled-coil peptide modules. Science 2013, 340, 595–599. [Google Scholar] [CrossRef]
- Nakase, I.; Okumura, S.; Tanaka, G.; Osaki, K.; Imanishi, M.; Futaki, S. Signal transduction using an artificial receptor system that undergoes dimerization upon addition of a bivalent leucine-zipper ligand. Angew. Chem. Int. Ed. 2012, 51, 7464–7467. [Google Scholar] [CrossRef]
- Thomas, F.; Boyle, A.L.; Burton, A.J.; Woolfson, D.N. A set of de novo designed parallel heterodimeric coiled coils with quantified dissociation constants in the micromolar to sub-nanomolar regime. J. Am. Chem. Soc. 2013, 135, 5161–5166. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Brown, I.R.; Juodeikis, R.; Frank, S.; Warren, M.J. Employing bacterial microcompartment technology to engineer a shell-free enzyme-aggregate for enhanced 1,2-propanediol production in Escherichia coli. Metab. Eng. 2016, 36, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leng, L.; Islam, M.K.; Liu, F.; Lin, C.S.; Leu, S.Y. Substrate-related factors affecting cellulosome-induced hydrolysis for lignocellulose valorization. Int. J. Mol. Sci. 2019, 20, 3354. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Kurosaki, M.; Nihei, S.; Hasegawa, H.; Shinoda, S.; Haruki, M.; Hirano, N. Enzymatic diversity of the Clostridium thermocellum cellulosome is crucial for the degradation of crystalline cellulose and plant biomass. Sci. Rep. 2016, 6, 35709. [Google Scholar] [CrossRef] [Green Version]
- Fontes, C.; Gilbert, H.J. Cellulosomes: Highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 2010, 79, 655–681. [Google Scholar] [CrossRef]
- Barth, A.; Hendrix, J.; Fried, D.; Barak, Y.; Bayer, E.A.; Lamb, D.C. Dynamic interactions of type I cohesin modules fine-tune the structure of the cellulosome of Clostridium thermocellum. Proc. Natl. Acad. Sci. USA 2018, 115, 11274–11283. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.H.; Zhang, Z.J.; Yu, X.Y.; Xue, Y.X.; Tan, T.W. Self-surface assembly of cellulosomes with two miniscaffoldins on Saccharomyces cerevisiae for cellulosic ethanol production. Proc. Natl. Acad. Sci. USA 2012, 109, 13260–13265. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.H.; Zhang, Z.J.; Yu, X.Y.; Xue, Y.X.; Wang, M.M.; Tan, T.W. In vitro assembly of minicellulosomes with two scaffoldins on the yeast cell surface for cellulose saccharification and bioethanol production. Process Biochem. 2013, 48, 430–437. [Google Scholar] [CrossRef]
- Kostas, T.; Sylvie, S.; Pierre, B. Interaction of the duplicated segment carried by Clostridium thermocellum cellulases with cellulosome components. FEBS Lett. 1991, 291, 185–188. [Google Scholar]
- Chen, C.; Yang, H.W.; Xuan, J.S. Resonance assignments of a cellulosomal double-dockerin from Clostridium thermocellum. Biomol. NMR Assign. 2019, 13, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.L.; Dias, F.; Prates, J.; Nagy, T.; Gilbert, H.J.; Davies, G.J.; Ferreira, L.; Romao, M.J.; Fontes, C. Cellulosome assembly revealed by the crystal structure of the cohesin-dockerin complex. Proc. Natl. Acad. Sci. USA 2003, 100, 13809–13814. [Google Scholar] [CrossRef] [Green Version]
- Betsy, L.L.; Brian, F.V.; William, M.W. Secondary structure and calcium-induced folding of the Clostridium thermocellum dockerin domain determined by NMR spectroscopy. Arch. Biochem. Biophys. 2000, 379, 237–244. [Google Scholar]
- Choi, S.K.; Ljungdah, L.G. Structural role of calcium for the organization of the cellulosome of Clostridium thermocellum. Biochemistry 1996, 35, 4906–4910. [Google Scholar] [CrossRef]
- Bule, P.; Cameron, K.; Prates, J.A.M.; Ferreira, L.M.A.; Smith, S.P.; Gilbert, H.J.; Bayer, E.A.; Najmudin, S.; Fontes, C.M.G.A.; Alves, V.D. Structure-function analyses generate novel specificities to assemble the components of multienzyme bacterial cellulosome complexes. J. Biol. Chem. 2018, 293, 4201–4212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Ding, S.Y.; Brunecky, R.; Bomble, Y.J.; Himmel, M.E.; Baker, J.O. Improving activity of minicellulosomes by integration of intra- and intermolecular synergies. Biotechnol. Biofuels 2013, 6, 126. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Sode, K.; Tamiya, E.; Karube, I.J.M. Enzymatic hydrolysis of cristalline cellulose in organic solvent. Metallomics 2011, 3, 656–661. [Google Scholar]
- Carvalho, A.L.; Dias, F.; Nagy, T.; Prates, J.; Proctor, M.R.; Smith, N.; Bayer, E.A.; Davies, G.J.; Ferreira, L.; Romao, M.J. Evidence for a dual binding mode of dockerin modules to cohesins. Proc. Natl. Acad. Sci. USA 2007, 104, 3089–3094. [Google Scholar] [CrossRef] [Green Version]
- Irina, G.; Margarita, M.; Elena, Z. New Kunitz-type HCRG polypeptides from the sea anemone Heteractis crispa. Mar. Drugs 2015, 13, 6038–6063. [Google Scholar]
- Morell, M.; Czihal, P.; Hoffmann, R.; Otvos, L.; Aviles, F.X.; Ventura, S. Monitoring the interference of protein-protein interactions in vivo by bimolecular fluorescence complementation: The DnaK case. Proteomics 2008, 8, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Espargaro, A.; Aviles, F.X.; Ventura, S. Detection of transient protein-protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 2007, 7, 1023–1036. [Google Scholar] [CrossRef]
- Parvatiyar, K.; Zhang, Z.; Teles, R.M.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ren, X.; Wang, R.; Su, J.; Wang, F. Structural characteristics and function of a new kind of thermostable trehalose synthase from Thermobaculum terrenum. J. Agric. Food Chem. 2017, 65, 7726–7735. [Google Scholar] [CrossRef]
- Xu, J.; Han, M.; Ren, X. Modification of aspartokinase III and dihydrodipicolinate synthetase increases the production of l-lysine in Escherichia coli. Biochem. Eng. J. 2016, 114, 79–86. [Google Scholar] [CrossRef]
- Chowdhury, C.; Sinha, S.; Chun, S.; Yeates, T.O.; Bobik, T.A. Diverse bacterial microcompartment organelles. Proc. Natl. Acad. Sci. USA 2014, 78, 438–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, B.P.; Chowdhury, C.; Bobik, T.A. The N terminus of the PduB protein binds the protein shell of the Pdu microcompartment to its enzymatic core. J. Bacteriol. 2017, 199, e00785-16. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Simpson, D.M.; Wenner, N.; Brownridge, P.; Harman, V.M.; Hinton, J.C.D.; Beynon, R.J.; Liu, L.N. Decoding the stoichiometric composition and organisation of bacterial metabolosomes. Nat. Commun. 2020, 11, 1976. [Google Scholar] [CrossRef] [Green Version]
- Kerfeld, C.A.; Melnicki, M.R. Assembly, function and evolution of cyanobacterial carboxysomes. Curr. Opin. Plant Biol. 2016, 31, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Rae, B.D.; Long, B.M.; Badger, M.R.; Price, G.D. Functions, compositions, and evolution of the two types of carboxysomes: Polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteria. Microbiol. Mol. Biol. Rev. 2013, 77, 357–379. [Google Scholar] [CrossRef] [Green Version]
- Amelung, S.; Nerlich, A.; Rohde, M.; Spellerberg, B.; Cole, J.N.; Nizet, V.; Chhatwal, G.S.; Talay, S.R. The FbaB-type fibronectin-binding protein of Streptococcus pyogenes promotes specific invasion into endothelial cells. Cell. Microbiol. 2011, 13, 1200–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, R.M.; Bjornsson, R.; McMahon, S.A.; Schomburg, B.; Braithwaite, V.; Buhl, M.; Naismith, J.H.; Schwarz-Linek, U. NMR spectroscopic and theoretical analysis of a spontaneously formed Lys-Asp isopeptide bond. Angew. Chem. Int. Ed. 2010, 49, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-display: Decoration of virus-like particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef]
- Chen, A.Y.; Deng, Z.T.; Billings, A.N.; Seker, U.O.S.; Lu, M.Y.; Citorik, R.J.; Zakeri, B.; Lu, T.K. Synthesis and patterning of tunable multiscale materials with engineered cells. Nat. Mater. 2014, 13, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Veggiani, G.; Zakeri, B.; Howarth, M. Superglue from bacteria: Unbreakable bridges for protein nanotechnology. Trends Biotechnol. 2014, 32, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Wang, X.; Zhang, D.; Wu, S.; Zhang, G. Enhanced thermal stability of lichenase from Bacillus subtilis 168 by SpyTag/SpyCatcher-mediated spontaneous cyclization. Biotechnol. Biofuels 2016, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, C.; Chun, S.; Pang, A.; Sawaya, M.R.; Sinha, S.; Yeates, T.O.; Bobik, T.A. Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc. Natl. Acad. Sci. USA 2015, 112, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, N.W.; Ikonomova, S.P.; Lee, M.S.; Raeder, H.W.; Tullman-Ercek, D. Self-assembling shell proteins PduA and PduJ have essential and redundant roles in bacterial microcompartment assembly. J. Mol. Biol. 2021, 433, 166721. [Google Scholar] [CrossRef]
- Pang, A.; Liang, M.; Prentice, M.B.; Pickersgill, R.W. Substrate channels revealed in the trimeric Lactobacillus reuteri bacterial microcompartment shell protein PduB. Acta Crystallogr. 2012, 68, 1642–1652. [Google Scholar]
- Lee, M.J.; Mantell, J.; Hodgson, L.; Alibhai, D.; Fletcher, J.M.; Brown, I.R.; Frank, S.; Xue, W.F.; Verkade, P.; Woolfson, D.N.; et al. Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat. Chem. Biol. 2018, 14, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Young, E.J.; Burton, R.; Mahalik, J.P.; Sumpter, B.G.; Fuentes-Cabrera, M.; Kerfeld, C.A.; Ducat, D.C. Engineering the bacterial microcompartment domain for molecular scaffolding applications. Front. Microbiol. 2017, 8, 1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, N.; Borne, R.; Fierobe, H.P. Cell-surface exposure of a hybrid 3-cohesin scaffoldin allowing the functionalization of Escherichia coli envelope. Biotechnol. Bioeng. 2020, 117, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.L.; Oh, J.; Singh, S.; Chen, R.; Chen, W. Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl. Environ. Microbiol. 2009, 75, 6087–6093. [Google Scholar] [CrossRef] [Green Version]
- Fierobe, H.P.; Bayer, E.A.; Tardif, C.; Czjzek, M.; Mechaly, A.; Bélaïch, A.; Lamed, R.; Shoham, Y.; Bélaïch, J.P. Degradation of cellulose substrates by cellulosome chimeras: Substrate targeting versus proximity of enzyme components. J. Biol. Chem. 2002, 277, 49621–49630. [Google Scholar] [CrossRef]
- Fierobe, H.P.; Mingardon, F.; Mechaly, A.; Bélaïch, A.; Rincon, M.T.; Pagès, S.; Lamed, R.; Tardif, C.; Bélaïch, J.P.; Bayer, E.A. Action of designer cellulosomes on homogeneous versus complex substrates-controlled incorporation of three distinct enzymes into a defined trifunctional scaffoldin. J. Biol. Chem. 2005, 280, 16325–16334. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.; Kuroda, K.; Ueda, M. Proximity effect among cellulose-degrading enzymes displayed on the Saccharomyces cerevisiae cell surface. Appl. Environ. Microbiol. 2015, 81, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Arsov, A.; Petrov, K.; Petrova, P. Enhanced activity by genetic complementarity: Heterologous secretion of clostridial cellulases by Bacillus licheniformis and Bacillus velezensis. Molecules 2021, 26, 5625. [Google Scholar] [CrossRef]
- Anandharaj, M.; Lin, Y.J.; Rani, R.P.; Nadendla, E.K.; Ho, M.C.; Huang, C.C.; Cheng, J.F.; Chang, J.J.; Li, W.H. Constructing a yeast to express the largest cellulosome complex on the cell surface. Proc. Natl. Acad. Sci. USA 2020, 117, 2385–2394. [Google Scholar] [CrossRef]
- Bule, P.; Alves, V.D.; Leitao, A.; Ferreira, L.M.A.; Bayer, E.A.; Smith, S.P.; Gilbert, H.J.; Najmudin, S.; Fontes, C. Single binding mode integration of hemicellulose-degrading enzymes via adaptor scaffoldins in Ruminococcus flavefaciens cellulosome. J. Biol. Chem. 2016, 291, 26658–26669. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.D.; Lee, J.E.; Kim, S.J.; Kim, S.W.; Han, S.O. Analysis of selective, high protein-protein binding interaction of cohesin-dockerin complex using biosensing methods. Biosens. Bioelectron. 2012, 35, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.Y.; Si, T.; Ang, E.L.; Zhao, H.M. Engineered pentafunctional minicellulosome for simultaneous saccharification and ethanol fermentation in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2014, 80, 6677–6684. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, I.R.; Yaniv, O.; de Ora, L.O.; Muñoz-Gutiérrez, I.; Hershko, A.; Livnah, O.; Bayer, E.A.; Borovok, I.; Frolow, F.; Lamed, R.; et al. Distinctive ligand-binding specificities of tandem PA14 biomass-sensory elements from Clostridium thermocellum and Clostridium clariflavum. Proteins Struct. Funct. Bioinform. 2019, 87, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnoo, M.; Cazade, P.-A.; Orlowski, A.; Chwastyk, M.; Liu, H.; Ta, D.T.; Cieplak, M.; Nash, M.; Thompson, D. Steered molecular dynamics simulations reveal the role of Ca2+ in regulating mechanostability of cellulose-binding proteins. Phys. Chem. Chem. Phys. 2018, 20, 22674–22680. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Iii, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2010, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
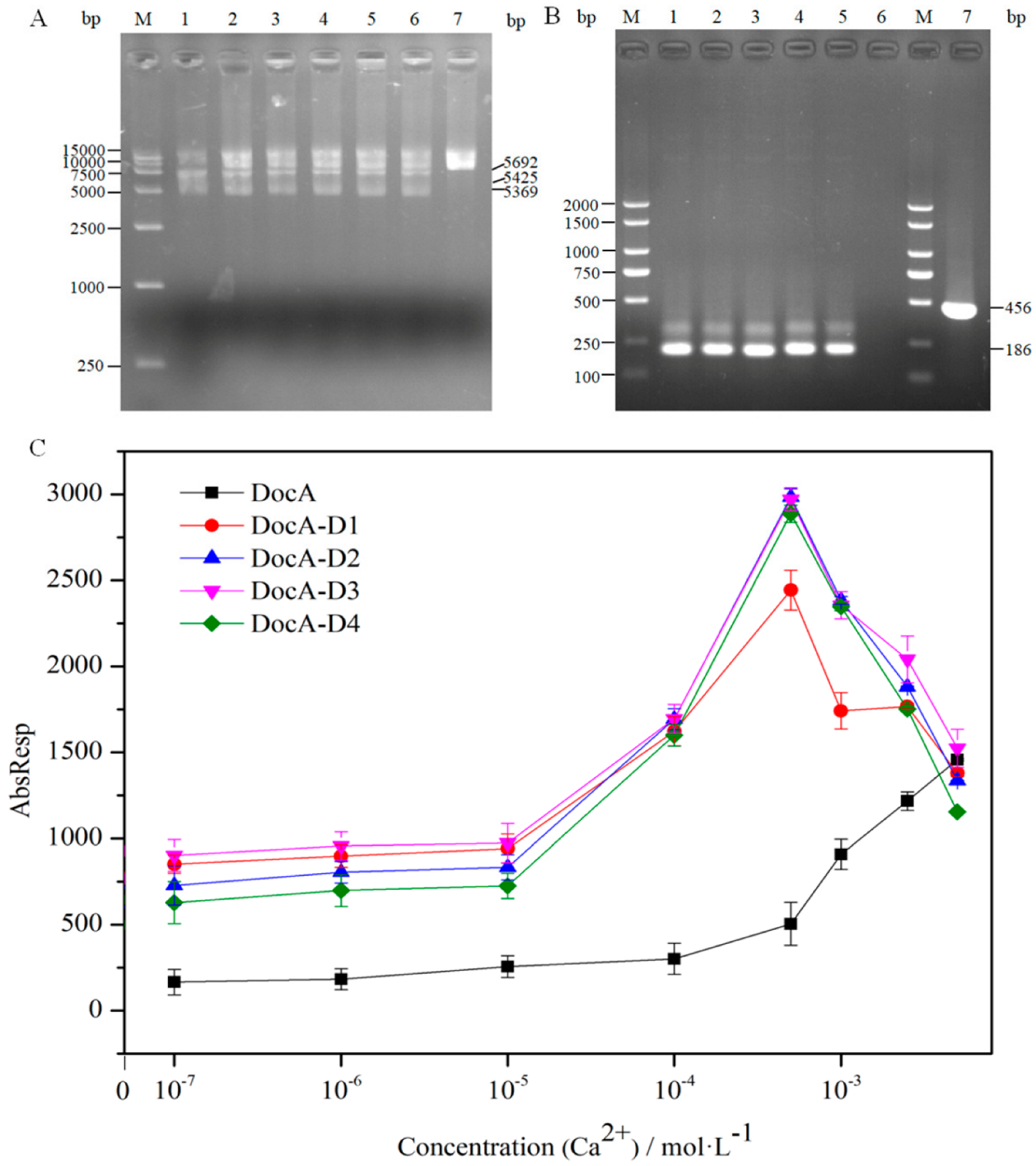{kind=link}
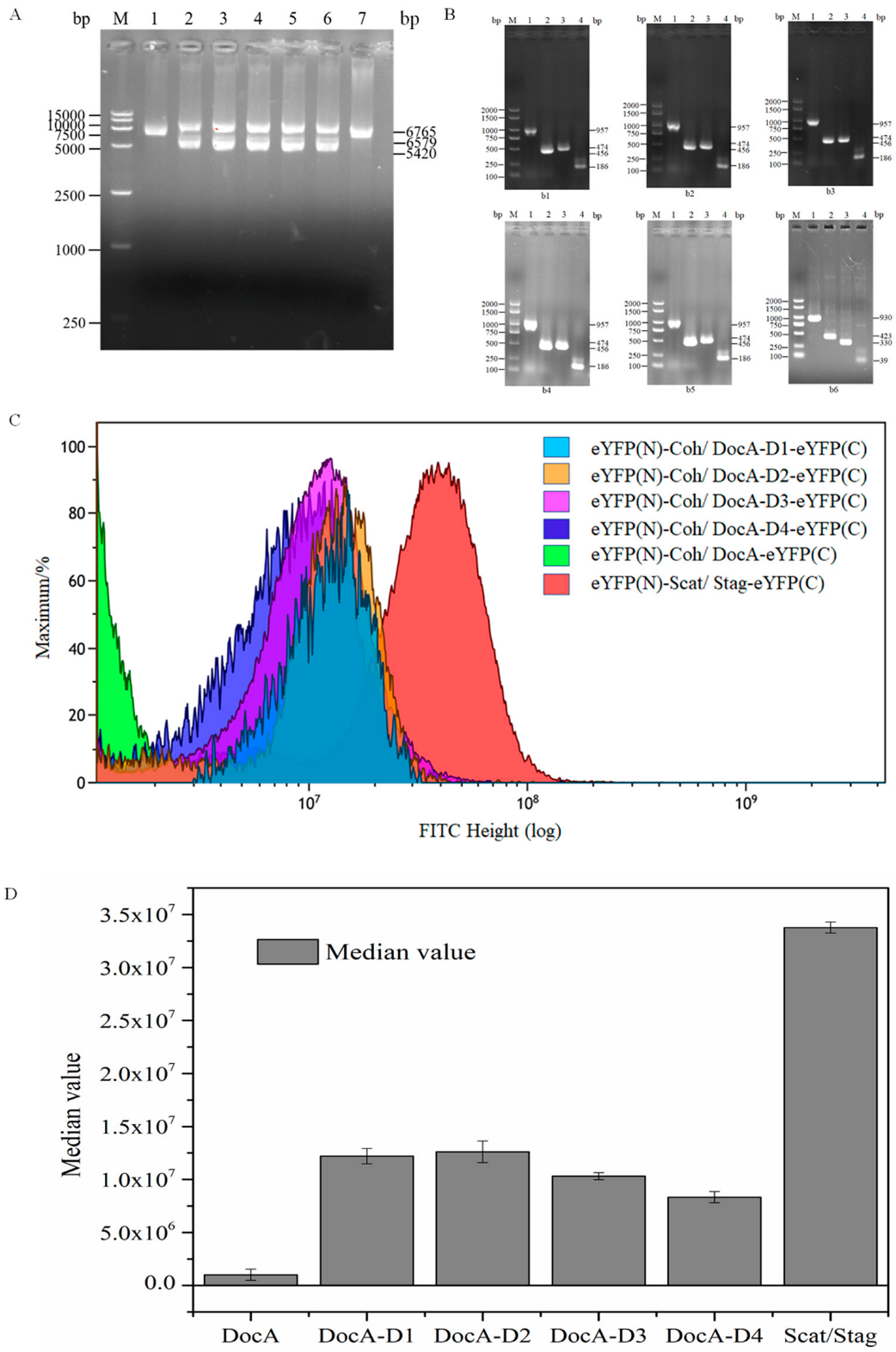{kind=link}
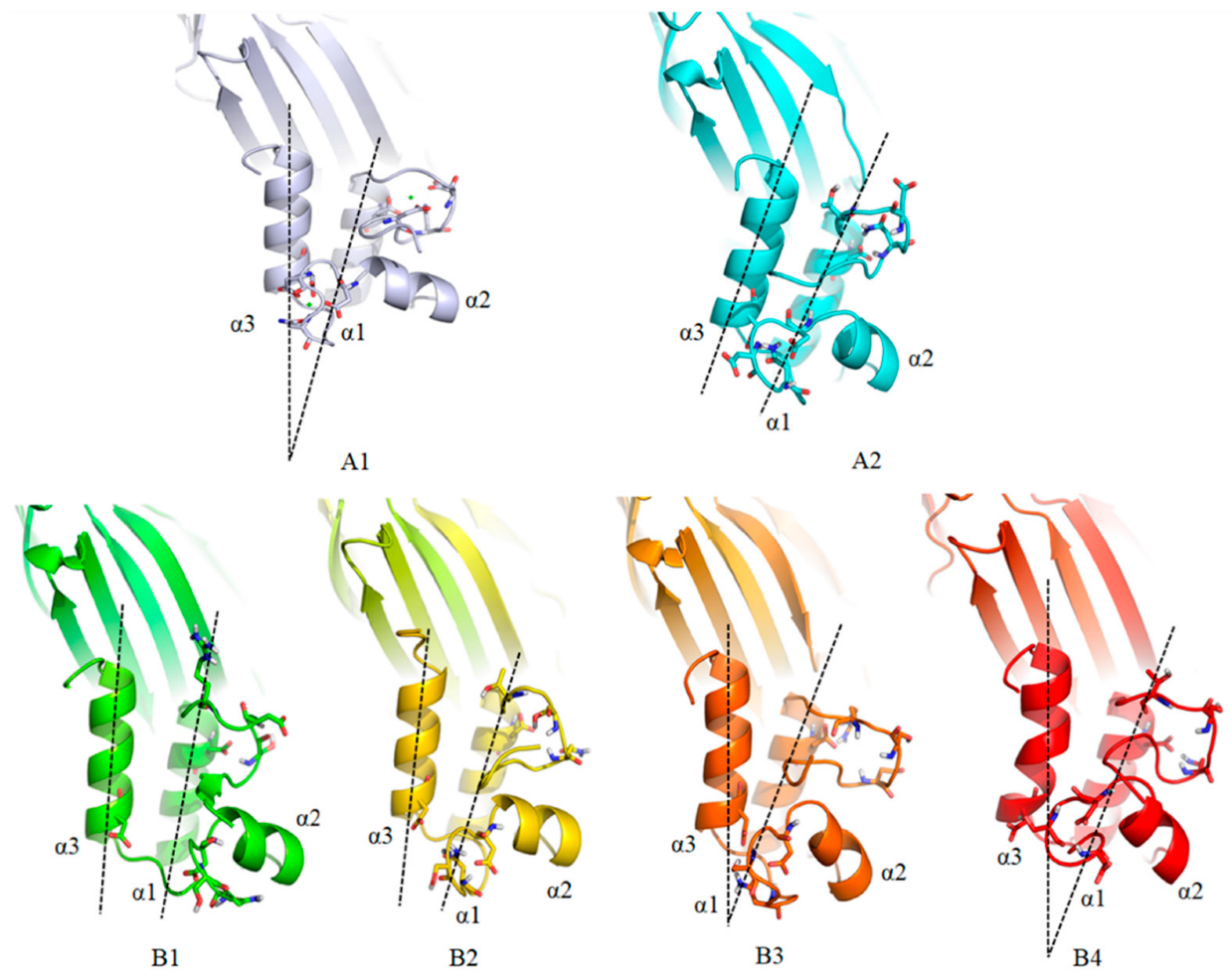{kind=link}
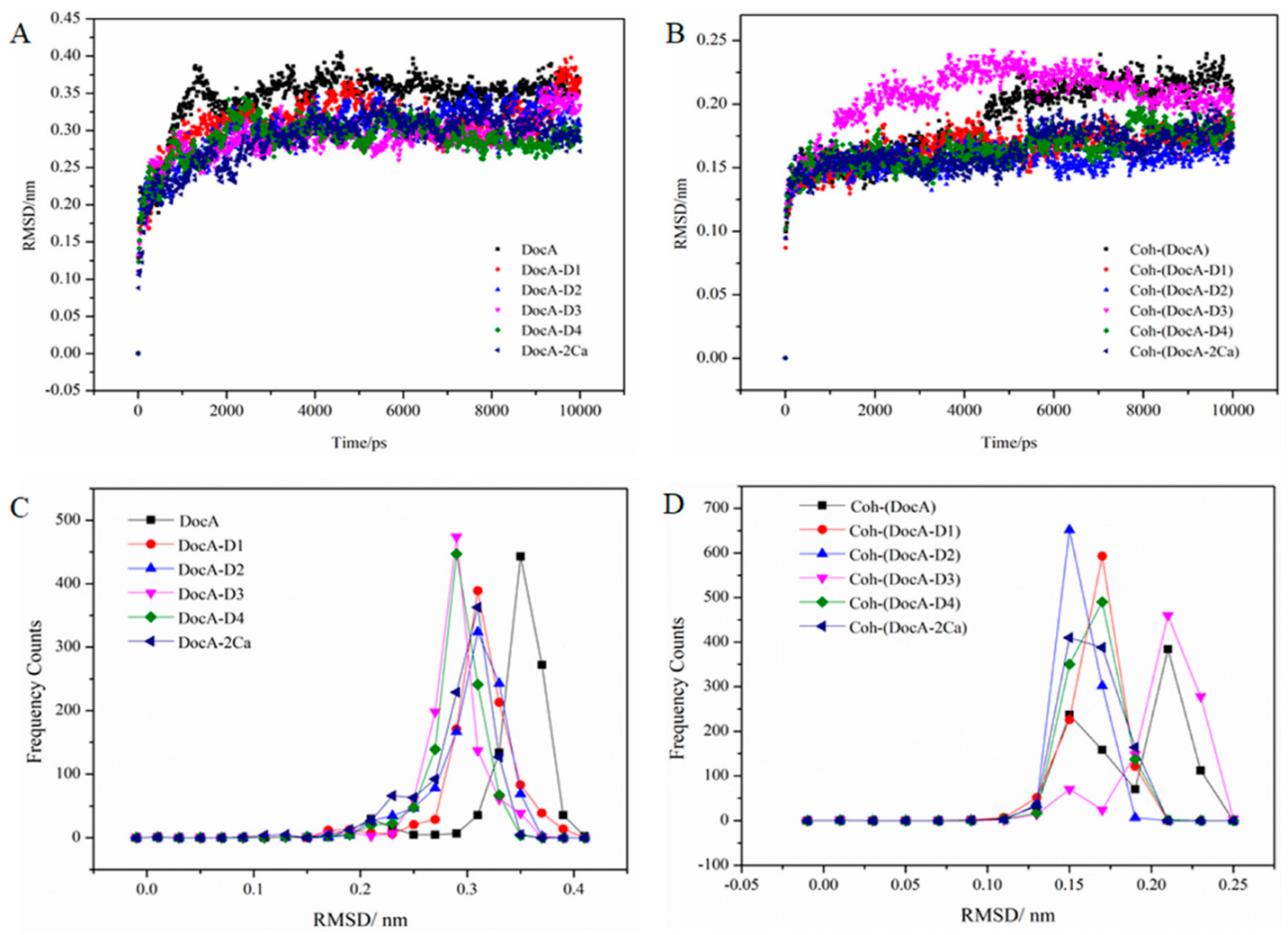{kind=link}
{kind=link}
{kind=link}
| Strains/Plasmids | Relevant Genotype | Source |
|---|---|---|
| Strains | ||
| E. coli DH5α | Amplification and extraction of plasmids | Vazyme |
| E. coli BL21(DE3) | Protein expression and extraction | Vazyme |
| E. coli QDE | L-lysine fermentation; ΔldcC, ΔamiD, ΔLYP1, icd-D410E, pykA-G168D | This study |
| Plasmids | ||
| pET-28a(+)-DocA | Expression of dockerin protein DocA | This study |
| pET-28a(+)-Coh | Expression of adhesin protein Coh | This study |
| pET-28a(+)-DocA-D1 | Expression of protein DocA-D1 | This study |
| pET-28a(+)-DocA-D2 | Expression of protein DocA-D2 | This study |
| pET-28a(+)-DocA-D3 | Expression of protein DocA-D3 | This study |
| pET-28a(+)-DocA-D4 | Expression of protein DocA-D4 | This study |
| pETDuet-1-eYFP(N)-Coh/DocA-eYFP(C) | BiFC expression protein DocA | This study |
| pETDuet-1-eYFP(N)-Coh/DocA-D1-eYFP(C) | BiFC expression protein DocA-D1 | This study |
| pETDuet-1-eYFP(N)-Coh/DocA-D2-eYFP(C) | BiFC expression protein DocA-D2 | This study |
| pETDuet-1-eYFP(N)-Coh/DocA-D3-eYFP(C) | BiFC expression protein DocA-D3 | This study |
| pETDuet-1-eYFP(N)-Coh/DocA-D4-eYFP(C) | BiFC expression protein DocA-D4 | This study |
| pETDuet-1-eYFP(N)-Scat/Stag-eYFP(C) | BiFC expression protein SpyCatcher/SpyTag | This study |
| pETDuet-1-aspC-Coh/DocA-D3-lysC | Intracellular assembly fermented L-lysine | This study |
| Protein | Primer Name | Sequence (5′→3′) |
|---|---|---|
| DocA-D1 | D1-F1 | TGGGTGACGTG tct GGTGACGGT cgt ATTAATA |
| D1-R1 | AGATCGGTGCTATTAAT acg ACCGTCACC aga CAC | |
| D1-F2 | AAAGCCCGTGCCGAT accagcaat AATGGC acc ATTAATG | |
| D1-R2 | AGAACATCGGCGGCATTAAT ggt GCCATT attgctggt ATCG | |
| DocA-D2 | D2-F1 | AATGGT agc GGTACCATTAATAGCA |
| D2-R1 | TACC gct ACCATTCACGTCACCCAGCA | |
| D2-F2 | CGAT accagcaat AATGGC acc ATTAATGCCGCCGATGTTCT | |
| D2-R2 | ATTAAT ggt GCCATT attgctggt ATCGGCACGGGCTTTGGCA | |
| DocA-D3 | D3-F1 | GTG gat GGT agc GGT cgt ATTAATAGCACCGAT |
| D3-R1 | TTAAT acg ACC gct ACC atc CACGTCACCCAGCA | |
| D3-F2 | CGAT accagcaat AATGGC acc ATTAATGCCGCCGATGTTCT | |
| D3-R2 | ATTAAT ggt GCCATT attgctggt ATCGGCACGGGCTTTGGCA | |
| DocA-D4 | D4-F1 | AATGGT agc GGTACCATTAATAGCA |
| D4-R1 | GTACC gct ACCATTCACGTCACCCAG | |
| D4-F2 | ATGTG agc AAA gat GGCAGCATTAATGCCGCCGAT | |
| D4-R2 | TGCC atc TTT gct CACATCGGCACGGGCTTTGGCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Xue, L.; Wang, Z.; Du, P.; Li, P.; Su, J.; Xiao, J.; Wang, M.; Wang, J.; Wang, R. Improving the Synthesis Efficiency of Amino Acids Such as L-Lysine by Assembling Artificial Cellulosome Elements Dockerin Protein In Vivo. Fermentation 2022, 8, 578. https://doi.org/10.3390/fermentation8110578
Li N, Xue L, Wang Z, Du P, Li P, Su J, Xiao J, Wang M, Wang J, Wang R. Improving the Synthesis Efficiency of Amino Acids Such as L-Lysine by Assembling Artificial Cellulosome Elements Dockerin Protein In Vivo. Fermentation. 2022; 8(11):578. https://doi.org/10.3390/fermentation8110578
Chicago/Turabian StyleLi, Nan, Le Xue, Zirui Wang, Peng Du, Piwu Li, Jing Su, Jing Xiao, Min Wang, Junqing Wang, and Ruiming Wang. 2022. "Improving the Synthesis Efficiency of Amino Acids Such as L-Lysine by Assembling Artificial Cellulosome Elements Dockerin Protein In Vivo" Fermentation 8, no. 11: 578. https://doi.org/10.3390/fermentation8110578
APA StyleLi, N., Xue, L., Wang, Z., Du, P., Li, P., Su, J., Xiao, J., Wang, M., Wang, J., & Wang, R. (2022). Improving the Synthesis Efficiency of Amino Acids Such as L-Lysine by Assembling Artificial Cellulosome Elements Dockerin Protein In Vivo. Fermentation, 8(11), 578. https://doi.org/10.3390/fermentation8110578