
A CRISPR-Cas12a-Based Assay for Efficient Quantification of Lactobacillus panis in Chinese Baijiu Brewing Microbiome

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Instruments
2.2. Preparation of Target Nucleic Acids
2.3. Preparation of crRNA and ssDNA Probes
2.4. Overview of the Detection Process
2.5. Verification of the CRISPR-Cas12a-Mediated Trans-Cleavage Fluorescent Reporter System (CQAOB)
2.6. Optimization of CQAOB
2.7. Analysis of the Detection Capability of CQAOB
2.8. Application of CQAOB Detection
2.9. Data Analysis
3. Results and Discussion
3.1. Workflow of the CQAOB System
3.2. Construction of a Lactobacillus panis DNA Fluorescence Reporter System Based on Cas12a
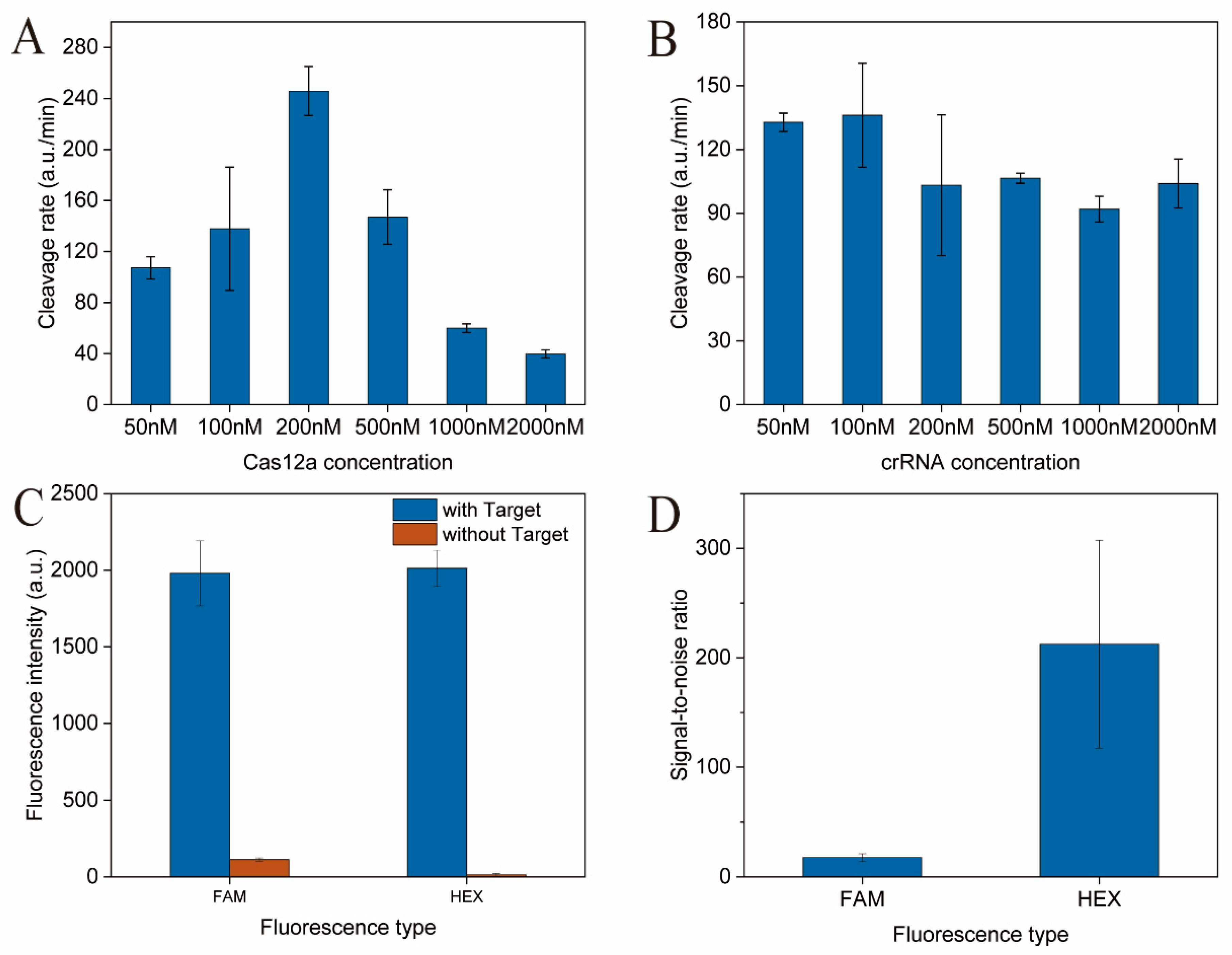
3.3. Optimization of the CRISPR-Cas12a Target DNA Detection System for L. panis
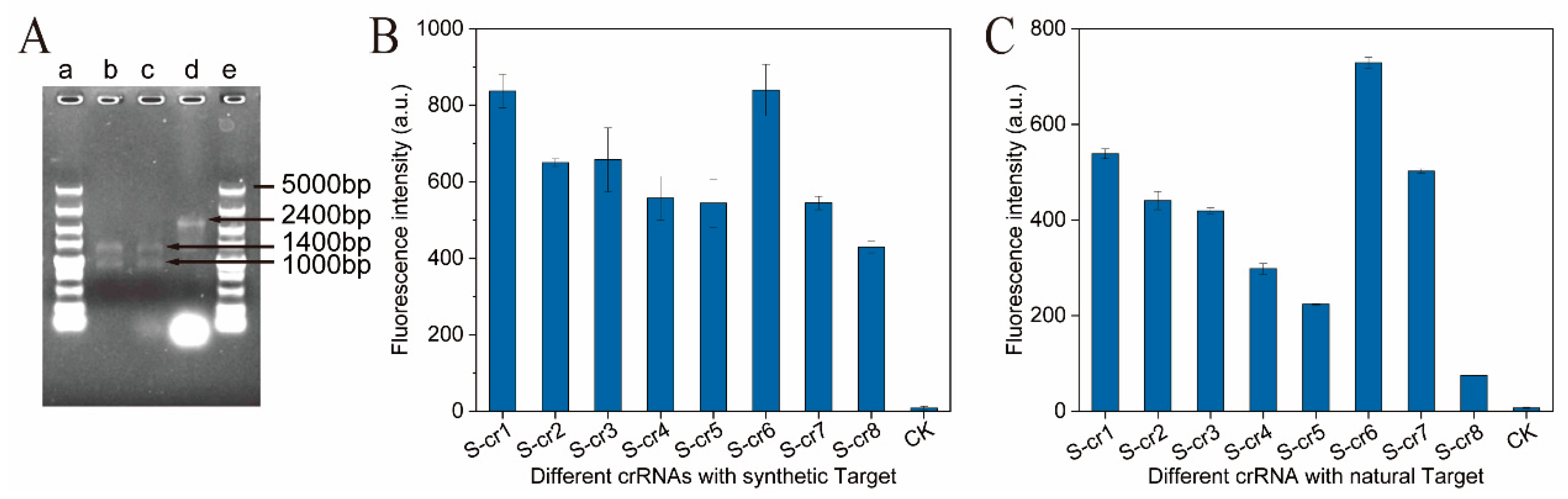
3.4. Analysis of the Specificity and Detection Range of the CRISPR-Cas12a Target DNA Detection System for L. panis
3.5. Consistency Assessment and Detection in Fermented Grains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, K. The Flavor of Moutai and Its Technological Characteristics. Food Mach. 1988, 1, 12–14, 16. [Google Scholar]
- Yang, F.; Chen, L.; Liu, Y.; Li, J.; Wang, L.; Chen, J. Identification of Microorganisms Producing Lactic Acid During Solid-State Fermentation of Maotai Flavour Liquor. J. Inst. Brew. 2019, 125, 171–177. [Google Scholar] [CrossRef]
- Zhou, H. Lactic Acid and Lactic Acid Bacteria Liquor-Making. Sci. Technol. 1995, 05, 11–14. [Google Scholar]
- Zeng, W.; Chen, F.; Zhan, X. The Production Technology of Lactic Acid and Its Research Progress. Chem. Ind. Eng. Prog. 2006, 7, 744–749. [Google Scholar]
- Li, W. Relationship Between Luzhou Flavor Liquor,Lactic Acid Bacteria,Lactic Acid and Ethyl Lactate. Liquor Mak. 2010, 37, 90–93. [Google Scholar]
- Yang, F.; Zhang, Q.; Liu, Y.; Li, J.; Wang, L.; Chen, J. Lactic Acid Biosynthesis Pathways and Important Genes of Lactobacillus panis L7 Isolated from the Chinese Liquor Brewing Microbiome. Food Biosci. 2020, 36, 100627. [Google Scholar] [CrossRef]
- Pejin, J.; Radosavljević, M.; Kocić-Tanackov, S.; Djukić-Vuković, A.; Mojović, L. Lactic Acid Fermentation of Brewer’s Spent Grain Hydrolysate by Lactobacillus rhamnosus with Yeast Extract Addition and pH Control. J. Inst. Brew. 2017, 123, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shu, Y.; Yang, J.; Gu, H.; Yang, Y. Research Progress of Quantitative Detection Methods of Lactic Acid Bacteria in Food. Sci. Technol. Food Ind. 2020, 41, 358–365. [Google Scholar]
- Fang, G.; Shan, L.; Li, H.; Wu, W.; Zhang, H. Study on the Counting Method of Active Lactic Acid Bacteria in Multi-Strain Yogurt. J. Microbiol. 2009, 29, 101–104. [Google Scholar]
- Li, Y.; Li, T. Study on the Methods of Affecting the Counting of Lactic Acid Bacteria Plate Colony. China Pract. Med. 2016, 11, 287–288. [Google Scholar]
- Du, R.; Wu, Q.; Xu, Y. Based on Three-Step Fluorescence Quantitative PCR Technology to Reveal the Distribution Characteristics of Lactobacillus sp in Liquor Brewing Systems in Different Producing Areas. J. Microbiol. 2020, 47, 1–12. [Google Scholar]
- Daranas, N.; Bonaterra, A.; Francés, J.; Cabrefiga, J.; Montesinos, E.; Badosa, E. Monitoring Viable Cells of the Biological Control Agent Lactobacillus plantarum PM411 in Aerial Plant Surfaces by Means of a Strain-Specific Viability Quantitative PCR Method. Appl. Environ. Microbiol. 2018, 84, e00107-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W. Fast Counting Method of Lactic Acid Bacteria Based on Flow Cytometry. China Food Saf. Mag. 2020, 21, 173. [Google Scholar]
- Dong, C. Application of Flow Cytometry in Rapid Detection of Bacteria. China Health Ind. 2014, 11, 194–195. [Google Scholar]
- Tourlousse, D.M.; Yoshiike, S.; Ohashi, A.; Matsukura, S.; Noda, N.; Sekiguchi, Y. Synthetic Spike-In Standards for High-Throughput 16S RRNA Gene Amplicon Sequencing. Nucleic Acids Res. 2017, 45, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Wu, Q.; Xu, Y. Chinese Liquor Fermentation: Identification of Key Flavor-Producing Lactobacillus spp. by Quantitative Profiling with Indigenous Internal Standards. Appl. Environ. Microbiol. 2020, 86, e00456-20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a Target Binding Unleashes Indiscriminate Single-Stranded DNase Activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 Proteins: Structure, Function and Implications for Genome Editing. Cell Biosci. 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.A.; Prakash, T.P.; Cleveland, D.W.; Bennett, C.F.; Rahdar, M. Chemically Modified Cpf1-CRISPR RNAs Mediate Efficient Genome Editing in Mammalian Cells. Mol. Ther. 2018, 26, 1228–1240. [Google Scholar] [CrossRef] [Green Version]
- Fozouni, P.; Son, S.; Díaz de León Derby, M.D.; Knott, G.J.; Gray, C.N.; D’Ambrosio, M.V.; Zhao, C.; Switz, N.A.; Kumar, G.R.; Stephens, S.I.; et al. Amplification-Free Detection of SARS-CoV-2 with CRISPR-Cas13a and Mobile Phone Microscopy. Cell 2021, 184, 323–333.e9. [Google Scholar] [CrossRef]
- Sun, Z.; Harris, H.M.B.; McCann, A.; Guo, C.; Argimón, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the Biotechnology Potential of Lactobacilli through Comparative Genomics of 213 Strains and Associated Genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef] [PubMed]
- Aman, R.; Mahas, A.; Marsic, T.; Hassan, N.; Mahfouz, M.M. Efficient, Rapid, and Sensitive Detection of Plant RNA Viruses with One-Pot RT-RPA–CRISPR/Cas12a Assay. Front. Microbiol. 2020, 11, 3277. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Sequences |
|---|---|
| crRNA transcription templates for gyrB | |
| g1 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATccaatgctgatgggaaccaaggt * |
| g2 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATccacagctgttcggcgtccattt |
| g3 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATttaagtacatcgaaagtgatgag |
| g4 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATtgcggccaatgatcacccacggc |
| g5 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATtcgtcgaaggggattccgccggt |
| g6 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATcggtggactccacggtgtggggg |
| g7 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATgaaacggtcttcacggttctgca |
| g8 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATtgtgacgaaatcaacgttgaggt |
| crRNA transcription templates for dsDNA cis-cleavage activity assays | |
| DNMT1 | GATCACTAATACGACTCACTATAGGAATTTCTACTCTTGTAGATCTgatggtccatgtctgttactc |
| DNMT2 | GATCACTAATACGACTCACTATAGGAATTTCTACTCTTGTAGATCTctgatggtccatgtctgttactcg |
| crRNA transcription templates for 16S rDNA | |
| S-cr1 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATggcattgcaaacttccatggtgt |
| S-cr2 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATcaatgcccaaagtcagtggccta |
| S-cr3 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATagagatttgcacaccctcgcggg |
| S-cr4 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATcacaccctcgcgggttagctgct |
| S-cr5 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATggatgggcccgcggtgcattagc |
| S-cr6 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATggctatcactttaggatgggccc |
| S-cr7 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATgtatcaaagatggtttcggctat |
| S-cr8 | GATCACTAATACGACTCACTATAGGTAATTTCTACTAAGTGTAGATgtagttatacggtattagcacct |
| Synthetic target DNAs for 16S rDNA | |
| S-ds1 | ACTTTGggcattgcaaacttccatggtgtGACGGGCGGTGTGTACAAGGC ** |
| S-ds2 | AGTTTGcaatgcccaaagtcagtggcctaACCATTTTGGAGGGAGCTGCC |
| S-ds3 | GGCTTTAagagatttgcacaccctcgcgggTTAGCTGCTCGTTGTACCGG |
| S-ds4 | AGATTTGcacaccctcgcgggttagctgctCGTTGTACCGGCCATTGTAG |
| S-ds5 | CACTTTAggatgggcccgcggtgcattagcTAGTTGGTAGGGTAACGGCC |
| S-ds6 | TGGTTTCggctatcactttaggatgggcccGCGGTGCATTAGCTAGTTGG |
| S-ds7 | GTTTTCgtatcaaagatggtttcggctatCACTTTAGGATGGGCCCGCGG |
| S-ds8 | GGTTTTCgtagttatacggtattagcacctGTTTCCAAATGTTATCCCCC |
| Gene expression elements | |
| T7 promoter | TAATACGACTCACTATAGG |
| Enhancer | GATCAC |
| ssDNA probes | |
| HEX5′-TTATT-3′BHQ1 *** | |
| FAM5′-TTATT-3′BHQ1 | |
| Primers | |
| DNMT-F | CACCAGTGAGACGGGCAAC |
| DNMT-R | ATTGCAGTTTCATTTGATGCTCGATG |
| 27F | AGTTTGATCMTGGCTCAG |
| 1492R | GGTTACCTTGTTACGACTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wang, M.; Yang, F.; Zhang, X.; Li, J.; Du, G.; Wang, L.; Chen, J. A CRISPR-Cas12a-Based Assay for Efficient Quantification of Lactobacillus panis in Chinese Baijiu Brewing Microbiome. Fermentation 2022, 8, 88. https://doi.org/10.3390/fermentation8020088
Liu Y, Wang M, Yang F, Zhang X, Li J, Du G, Wang L, Chen J. A CRISPR-Cas12a-Based Assay for Efficient Quantification of Lactobacillus panis in Chinese Baijiu Brewing Microbiome. Fermentation. 2022; 8(2):88. https://doi.org/10.3390/fermentation8020088
Chicago/Turabian StyleLiu, Yanfeng, Mengchuang Wang, Fan Yang, Xiaolong Zhang, Jianghua Li, Guocheng Du, Li Wang, and Jian Chen. 2022. "A CRISPR-Cas12a-Based Assay for Efficient Quantification of Lactobacillus panis in Chinese Baijiu Brewing Microbiome" Fermentation 8, no. 2: 88. https://doi.org/10.3390/fermentation8020088
APA StyleLiu, Y., Wang, M., Yang, F., Zhang, X., Li, J., Du, G., Wang, L., & Chen, J. (2022). A CRISPR-Cas12a-Based Assay for Efficient Quantification of Lactobacillus panis in Chinese Baijiu Brewing Microbiome. Fermentation, 8(2), 88. https://doi.org/10.3390/fermentation8020088