
Methodology for Analysis of Peptide Consumption by Yeast during Fermentation of Enzymatic Protein Hydrolysate Supplemented Synthetic Medium Using UPLC-IMS-HRMS


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Fermentation Media
2.2.1. BSA Hydrolysate
2.2.2. Synthetic Grape Must
2.3. Fermentation
2.4. Sample Preparation
2.5. Liquid Chromatography Mass Spectrometry
2.6. Data Processing
2.7. Data Analysis
3. Results
3.1. BSA Hydrolysate Composition
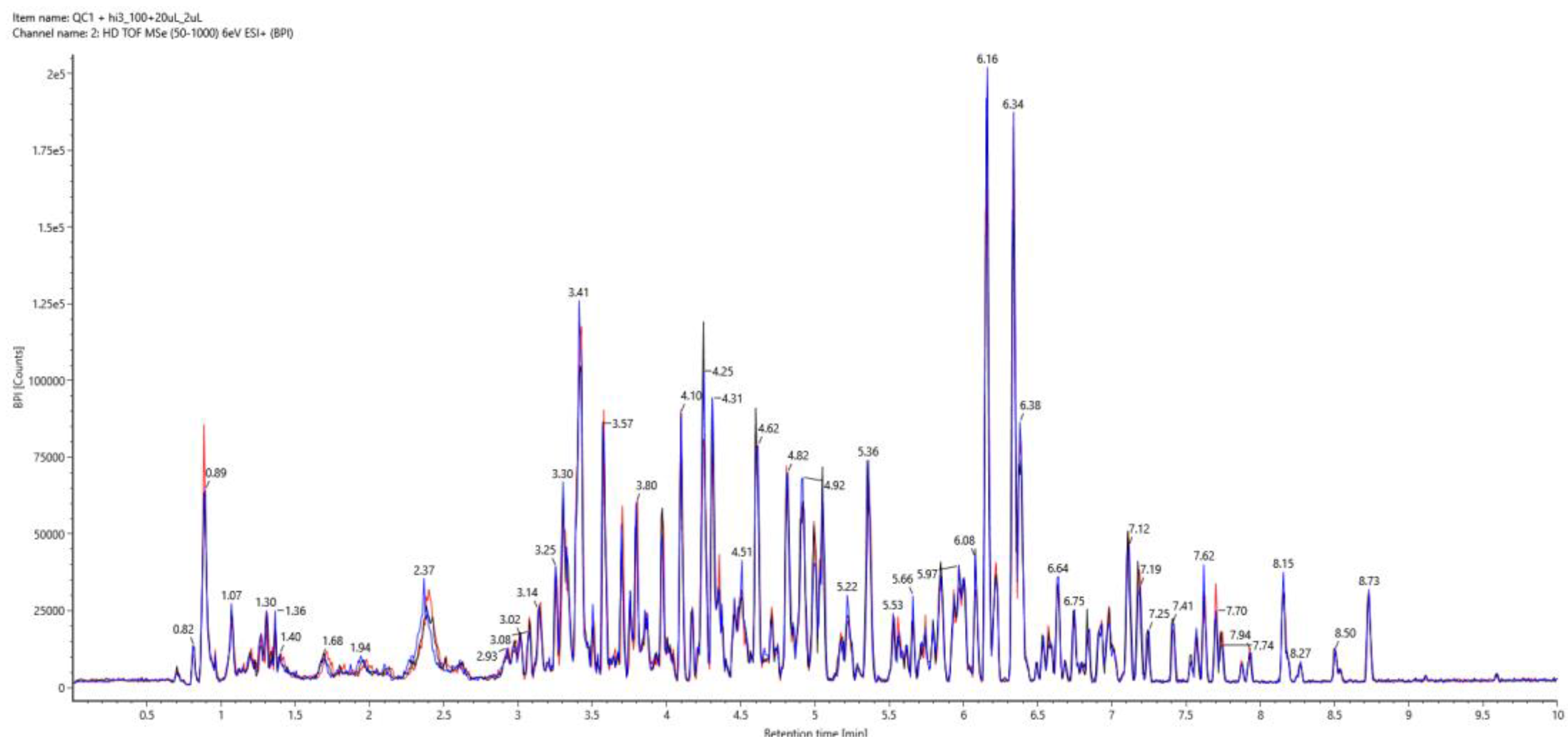
3.2. LC-MS Method Reproducibility
3.3. Fermentation and Peptide Uptake Kinetics
3.4. Untargeted Peptide Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Glucose Consumed (g⋅L−1) | Fructose Consumed (g⋅L−1) | Ethanol Produced (g⋅L−1) | |
|---|---|---|---|---|
| Lalvin ICV Opale 2.0™ | Control | 97.3 | 72.4 | 77.5 |
| BSA | 102.8 | 97.3 | 95.4 | |
| Lalvin Persy™ | Control | 97.4 | 76.8 | 81.4 |
| BSA | 104.4 | 101.2 | 95.7 | |
| Lalvin QA23™ | Control | 90.1 | 66.9 | 75.5 |
| BSA | 100.0 | 92.1 | 96.0 |
Appendix B
| Compound | Amount (mg⋅L−1) | Compound | Amount (mg⋅L−1) |
|---|---|---|---|
| Tryptophan | 38.7 | Alanine | 31.4 |
| Phenylalanine | 8.2 | Threonine | 16.4 |
| Isoleucine | 7.1 | Glutamic acid | 26.0 |
| Leucine | 10.5 | Aspartic acid | 9.6 |
| Valine | 9.6 | Glycine | 4.0 |
| Methionine | 6.8 | Arginine | 80.8 |
| Tyrosine | 4.0 | Glutamine | 109.1 |
| Lysine | 3.7 | Serine | 17.0 |
| Cysteine | 2.8 | Asparagine | 0.0 |
| Proline | 132.2 | Histidine | 7.1 |
| NH4CL | 102.5 |
Appendix C
| Strain | N from FAA (mg·L−1) | N from BAA (mg·L−1) | N from NH4 (mg·L−1) | N Total (mg·L−1) | |
|---|---|---|---|---|---|
| Lalvin ICV Opale 2.0™ | Control | 70.20 ± 0.07 | - | 18.08 ± 6.87 | 88.2 ± 6.87 |
| BSA | 77.44 ± 0.22 | 74.4 ± 2.12 | 22.49 ± 2.23 | 174.3 ± 3.08 | |
| Lalvin Persy™ | Control | 75.8 ± 0.17 | - | 26.96 ± 6.35 | 102.8 ± 6.65 |
| BSA | 72.0 ± 0.18 | 57.3 ± 0.53 | 25.41 ± 5.07 | 154.7 ± 5.10 | |
| Lalvin QA23™ | Control | 75.50 ± 0.20 | - | 25.13 ± 6.19 | 100.6 ± 6.49 |
| BSA | 73.79 ± 0.16 | 51.7 ± 5.87 | 25.41 ± 5.07 | 150.9 ± 7.76 |
Appendix D
| Peptide Candidate | Retention Time (min) | Observed m/z | Charge | CCS (Å2) | Average Original Intensity | RSD | Lalvin ICV Opale 2.0™ (Consumption, %) | Lalvin Persy™ (Consumption, %) | Lalvin QA23™ (Consumption, %) |
|---|---|---|---|---|---|---|---|---|---|
| AH | 2.68 | 227.1134 | 1 | 150.41 | 807 | 4.86 | 99.97 | 100.00 | 99.98 |
| AW | 5.40 | 276.1338 | 1 | 160.10 | 1914 | 5.03 | 100.00 | 100.00 | 100.00 |
| AY | 4.56 | 253.1177 | 1 | 157.67 | 1470 | 6.44 | 100.00 | 100.00 | 100.00 |
| FT | 3.72 | 267.1335 | 1 | 163.99 | 5487 | 3.53 | 100.00 | 100.00 | 100.00 |
| FW | 7.03 | 352.1652 | 1 | 156.33 | 2617 | 5.18 | 100.00 | 100.00 | 100.00 |
| FY | 5.38 | 329.1487 | 1 | 179.32 | 698 | 3.61 | 97.94 | 95.49 | 95.50 |
| JT | 3.18 | 233.1492 | 1 | 156.95 | 2875 | 4.95 | 99.99 | 100.00 | 100.00 |
| KQ | 2.39 | 275.1709 | 1 | 160.14 | 1164 | 2.32 | 99.99 | 99.82 | 99.87 |
| KT | 1.40 | 248.1601 | 1 | 156.18 | 2201 | 8.75 | 99.97 | 99.98 | 100.00 |
| LL | 5.84 | 245.1856 | 1 | 166.83 | 20,475 | 3.04 | 100.00 | 100.00 | 100.00 |
| LY | 4.88 | 295.1648 | 1 | 171.64 | 4360 | 3.64 | 100.00 | 100.00 | 100.00 |
| TF | 4.78 | 267.1335 | 1 | 160.50 | 1507 | 3.99 | 100.00 | 99.87 | 100.00 |
| TK | 2.51 | 248.1600 | 1 | 156.18 | 2070 | 2.46 | 100.00 | 100.00 | 100.00 |
| VE | 2.14 | 247.1285 | 1 | 154.51 | 6009 | 6.03 | 99.96 | 99.98 | 99.95 |
| VF | 5.50 | 265.1542 | 1 | 164.08 | 5044 | 4.39 | 100.00 | 100.00 | 100.00 |
| VN | 1.37 | 232.1288 | 1 | 150.14 | 1012 | 2.78 | 99.98 | 100.00 | 100.00 |
| VR | 3.30 | 274.1866 | 1 | 167.20 | 1039 | 5.77 | 100.00 | 100.00 | 100.00 |
| YE | 3.72 | 311.1229 | 1 | 172.79 | 1091 | 4.24 | 99.67 | 100.00 | 100.00 |
| YY | 4.55 | 345.1441 | 1 | 184.22 | 5445 | 5.16 | 100.00 | 100.00 | 100.00 |
| Peptide Candidate | Retention Time (min) | Observed m/z | Charge | CCS (Å2) | Average Original Intensity | %RSD | Lalvin ICV Opale 2.0™ (Consumption, %) | Lalvin Persy™ (Consumption, %) | Lalvin QA23™ (Consumption, %) |
|---|---|---|---|---|---|---|---|---|---|
| AEF | 5.27 | 366.1656 | 1 | 181.69 | 5873 | 4.08 | 100.00 | 100.00 | 100.00 |
| AWS | 4.68 | 363.1658 | 1 | 181.79 | 2470 | 4.50 | 100.00 | 100.00 | 100.00 |
| DAF | 5.21 | 352.1496 | 1 | 176.71 | 1100 | 2.70 | 100.00 | 100.00 | 100.00 |
| DLL | 5.76 | 360.2124 | 1 | 189.23 | 2953 | 6.90 | 100.00 | 100.00 | 100.00 |
| DQF | 4.88 | 409.1712 | 1 | 195.14 | 728 | 3.28 | 100.00 | 100.00 | 100.00 |
| ELT | 4.01 | 362.1919 | 1 | 187.32 | 3543 | 2.99 | 99.98 | 99.62 | 99.92 |
| FKD | 3.53 | 205.1072 | 2 | 249.17 | 7981 | 3.00 | 100.00 | 100.00 | 100.00 |
| FKG | 3.33 | 176.1043 | 2 | 242.20 | 2685 | 2.41 | 100.00 | 100.00 | 100.00 |
| FLG | 5.65 | 336.1910 | 1 | 182.71 | 734 | 3.52 | 100.00 | 100.00 | 100.00 |
| FQE | 4.32 | 423.1866 | 1 | 198.49 | 658 | 3.50 | 100.00 | 100.00 | 100.00 |
| FSA | 4.73 | 324.1549 | 1 | 174.09 | 3223 | 2.23 | 100.00 | 100.00 | 100.00 |
| FTF | 7.23 | 414.2020 | 1 | 194.99 | 3313 | 6.62 | 100.00 | 100.00 | 100.00 |
| FVE | 4.88 | 394.1970 | 1 | 190.00 | 3242 | 3.60 | 100.00 | 100.00 | 100.00 |
| GSF | 4.64 | 310.1391 | 1 | 167.50 | 1014 | 6.12 | 100.00 | 100.00 | 100.00 |
| IAE | 3.72 | 332.1811 | 1 | 175.60 | 2444 | 4.33 | 100.00 | 100.00 | 100.00 |
| IAF | 6.63 | 350.2071 | 1 | 180.40 | 5291 | 6.60 | 100.00 | 100.00 | 100.00 |
| IAH | 2.66 | 340.1973 | 1 | 182.57 | 1628 | 7.50 | 100.00 | 100.00 | 100.00 |
| IAR | 3.08 | 359.2396 | 1 | 185.57 | 2132 | 6.89 | 100.00 | 100.00 | 100.00 |
| ISL | 6.42 | 332.2170 | 1 | 182.85 | 933 | 5.48 | 100.00 | 100.00 | 100.00 |
| IVR | 3.31 | 194.1387 | 2 | 253.10 | 4260 | 4.53 | 99.92 | 99.99 | 99.80 |
| KIE | 3.26 | 195.1228 | 2 | 243.74 | 2444 | 6.39 | 99.61 | 99.86 | 99.84 |
| KQI | 2.40 | 194.6309 | 2 | 246.86 | 8652 | 6.83 | 100.00 | 100.00 | 100.00 |
| LAK | 2.50 | 331.2332 | 1 | 181.06 | 482 | 3.99 | 100.00 | 100.00 | 100.00 |
| LEE | 3.90 | 390.1876 | 1 | 188.27 | 1557 | 5.17 | 100.00 | 93.10 | 94.29 |
| LFT | 5.92 | 380.2172 | 1 | 190.43 | 706 | 3.45 | 100.00 | 100.00 | 100.00 |
| LLF | 8.00 | 392.2539 | 1 | 197.53 | 1399 | 5.91 | 100.00 | 100.00 | 100.00 |
| LSQ | 3.30 | 347.1919 | 1 | 178.69 | 3179 | 5.20 | 100.00 | 100.00 | 100.00 |
| LVE | 4.37 | 360.2126 | 1 | 183.71 | 18,437 | 5.06 | 100.00 | 100.00 | 100.00 |
| LVN | 3.77 | 345.2125 | 1 | 180.57 | 990 | 4.59 | 100.00 | 100.00 | 100.00 |
| LYE | 4.68 | 424.2076 | 1 | 196.58 | 9328 | 3.77 | 100.00 | 100.00 | 100.00 |
| LYY | 6.08 | 458.2284 | 1 | 207.03 | 3200 | 6.61 | 100.00 | 99.99 | 100.00 |
| SQY | 3.78 | 397.1714 | 1 | 186.22 | 3568 | 5.93 | 100.00 | 100.00 | 100.00 |
| SVL | 5.63 | 318.2013 | 1 | 176.11 | 265 | 7.45 | 100.00 | 100.00 | 99.94 |
| TEF | 5.23 | 396.1756 | 1 | 189.94 | 620 | 3.45 | 100.00 | 100.00 | 100.00 |
| TLV | 4.44 | 332.2174 | 1 | 181.03 | 1307 | 2.71 | 100.00 | 100.00 | 100.00 |
| VAF | 6.01 | 336.1915 | 1 | 175.46 | 10,184 | 2.70 | 100.00 | 99.96 | 100.00 |
| VNE | 3.12 | 361.1714 | 1 | 178.22 | 5809 | 4.62 | 100.00 | 100.00 | 100.00 |
| VTF | 5.96 | 366.2019 | 1 | 179.87 | 1740 | 6.69 | 100.00 | 100.00 | 100.00 |
| VTK | 1.40 | 347.2286 | 1 | 180.50 | 3469 | 6.77 | 100.00 | 100.00 | 100.00 |
| YEY | 5.06 | 474.1869 | 1 | 208.53 | 3085 | 5.50 | 99.98 | 100.00 | 99.99 |
| YNG | 3.07 | 353.1448 | 1 | 183.95 | 737 | 3.00 | 100.00 | 100.00 | 100.00 |
| Peptide Candidate | Retention Time (min) | Observed m/z | Charge | CCS (Å2) | Average Original Intensity | %RSD | Lalvin ICV Opale 2.0™ (Consumption, %) | Lalvin Persy™ (Consumption, %) | Lalvin QA23™ (Consumption, %) |
|---|---|---|---|---|---|---|---|---|---|
| ALVE | 5.13 | 431.2496 | 1 | 200.15 | 3448 | 7.14 | 100.00 | 98.07 | 99.34 |
| FAKT | 3.49 | 233.6360 | 2 | 266.06 | 2319 | 7.68 | 100.00 | 100.00 | 100.00 |
| FEGK | 5.44 | 269.1441 | 2 | 279.98 | 9697 | 5.25 | 74.05 | 46.42 | 96.43 |
| FEKL | 5.27 | 268.6570 | 2 | 276.78 | 1106 | 8.84 | 96.81 | 95.23 | 93.37 |
| FHAD | 3.66 | 245.1077 | 2 | 262.19 | 5008 | 5.24 | 100.00 | 100.00 | 99.99 |
| FKDL | 5.26 | 261.6491 | 2 | 277.18 | 3826 | 7.25 | 100.00 | 100.00 | 99.40 |
| FLGS | 5.62 | 423.2233 | 1 | 198.49 | 1214 | 1.92 | 100.00 | 100.00 | 100.00 |
| FSQY | 5.07 | 544.2400 | 1 | 222.40 | 5451 | 4.56 | 99.89 | 99.65 | 99.77 |
| GERA | 1.71 | 216.6131 | 2 | 254.59 | 646 | 5.90 | 84.37 | 71.04 | 78.31 |
| IETM | 5.11 | 493.2326 | 1 | 215.78 | 10,279 | 5.05 | 100.00 | 95.85 | 98.39 |
| IKQN | 1.91 | 251.6520 | 2 | 261.81 | 745 | 8.35 | 100.00 | 95.20 | 98.59 |
| ISSK | 1.99 | 217.6335 | 2 | 257.65 | 1951 | 10.49 | 100.00 | 99.71 | 99.07 |
| KDAF | 4.45 | 480.2451 | 1 | 204.55 | 4169 | 6.61 | 93.70 | 37.87 | 41.44 |
| LEKS | 2.52 | 238.6388 | 2 | 262.58 | 3371 | 5.37 | 100.00 | 100.00 | 99.97 |
| LGSF | 6.08 | 423.2236 | 1 | 196.61 | 7284 | 4.72 | 93.04 | 76.01 | 95.77 |
| LILN | 6.24 | 472.3125 | 1 | 218.28 | 2581 | 5.87 | 100.00 | 100.00 | 100.00 |
| LIVR | 4.82 | 250.6806 | 2 | 277.82 | 1165 | 3.78 | 75.70 | 49.87 | 50.76 |
| LLEK | 4.13 | 251.6648 | 2 | 271.33 | 4134 | 3.64 | 91.38 | 48.34 | 49.20 |
| LRET | 3.30 | 259.6495 | 2 | 267.68 | 915 | 7.21 | 100.00 | 100.00 | 100.00 |
| LTAD | 3.81 | 419.2132 | 1 | 194.85 | 2055 | 6.78 | 100.00 | 100.00 | 100.00 |
| LTEF | 6.28 | 509.2602 | 1 | 217.34 | 2520 | 6.27 | 100.00 | 100.00 | 100.00 |
| LVEL | 6.85 | 473.2970 | 1 | 214.35 | 7655 | 5.36 | 100.00 | 99.36 | 99.86 |
| LVNE | 4.15 | 474.2558 | 1 | 208.53 | 6118 | 6.06 | 99.95 | 99.71 | 99.98 |
| LVTD | 4.33 | 447.2446 | 1 | 201.61 | 8109 | 3.72 | 100.00 | 99.72 | 99.85 |
| MENF | 5.75 | 540.2122 | 1 | 222.49 | 2536 | 6.75 | 100.00 | 100.00 | 100.00 |
| TQTA | 2.58 | 420.2083 | 1 | 196.70 | 1160 | 6.33 | 100.00 | 100.00 | 100.00 |
| VASL | 2.93 | 195.1228 | 2 | 253.02 | 1457 | 5.55 | 99.77 | 99.68 | 99.85 |
| VEVS | 4.02 | 433.2284 | 1 | 198.21 | 2150 | 6.29 | 98.70 | 97.71 | 97.77 |
| VFDK | 4.02 | 254.6413 | 2 | 267.97 | 1976 | 5.98 | 69.07 | 35.82 | 53.99 |
| VSEK | 1.95 | 231.6310 | 2 | 259.87 | 2212 | 7.45 | 99.97 | 97.52 | 92.49 |
| VVST | 3.70 | 405.2340 | 1 | 191.53 | 3913 | 3.23 | 100.00 | 100.00 | 100.00 |
| Peptide Candidate | Retention Time (min) | Observed m/z | Charge | CCS (Å2) | Average Original Intensity | RSD | Lalvin ICV Opale 2.0™ (Consumption, %) | Lalvin Persy™ (Consumption, %) | Lalvin QA23™ (Consumption, %) |
|---|---|---|---|---|---|---|---|---|---|
| AFDEK | 3.91 | 305.1472 | 2 | 274.91 | 15,458 | 6.56 | 99.93 | 36.21 | 41.18 |
| AIPEN | 4.24 | 543.2772 | 1 | 218.50 | 8777 | 4.98 | 96.76 | 33.49 | 33.68 |
| ALVEL | 7.32 | 544.3338 | 1 | 230.33 | 912 | 7.41 | 99.99 | 40.31 | 48.36 |
| FDEKL | 5.50 | 326.1703 | 2 | 300.05 | 5931 | 5.12 | 100.00 | 58.69 | 61.54 |
| FLGSF | 7.55 | 570.2920 | 1 | 231.74 | 934 | 6.74 | 99.96 | 33.44 | 31.64 |
| FYAPE | 5.74 | 626.2814 | 1 | 244.72 | 2299 | 4.18 | 100.00 | 66.49 | 66.40 |
| GFQNA | 4.50 | 536.2458 | 1 | 220.62 | 1214 | 7.16 | 78.76 | 34.47 | 38.77 |
| KFWGK | 5.00 | 333.1909 | 2 | 296.41 | 878 | 4.19 | 100.00 | 99.98 | 100.00 |
| LAKEY | 4.23 | 312.1727 | 2 | 277.79 | 4602 | 2.52 | 100.00 | 61.22 | 91.98 |
| LFGDE | 6.00 | 580.2609 | 1 | 227.53 | 825 | 7.83 | 99.79 | 28.10 | 35.07 |
| LGEYG | 4.95 | 538.2504 | 1 | 218.61 | 831 | 3.51 | 90.99 | 32.33 | 35.78 |
| LILNR | 5.33 | 314.7094 | 2 | 287.40 | 797 | 7.07 | 99.75 | 30.78 | 47.93 |
| LPKIE | 5.42 | 300.1912 | 2 | 284.85 | 8843 | 6.91 | 100.00 | 60.78 | 99.87 |
| LVELL | 7.98 | 586.3803 | 1 | 241.49 | 707 | 5.49 | 90.22 | 33.34 | 64.02 |
| LVEVS | 5.21 | 546.3130 | 1 | 224.32 | 4239 | 0.63 | 99.26 | 39.13 | 54.87 |
| TAPEL | 6.26 | 592.2976 | 1 | 235.28 | 15,546 | 5.29 | 100.00 | 93.48 | 93.12 |
| TVFDK | 4.61 | 305.1650 | 2 | 284.60 | 2748 | 4.80 | 99.98 | 66.96 | 61.71 |
| VEVTK | 3.65 | 288.1730 | 2 | 275.74 | 7041 | 8.04 | 98.83 | 39.91 | 48.04 |
| VVSTQ | 3.53 | 533.2928 | 1 | 218.72 | 3638 | 5.90 | 98.97 | 43.60 | 46.92 |
| Peptide Candidate | Retention Time (min) | Observed m/z | Charge | CCS (Å2) | Average Original Intensity | %RSD | Lalvin ICV Opale 2.0™ (Consumption, %) | Lalvin Persy™ (Consumption, %) | Lalvin QA23™ (Consumption, %) |
|---|---|---|---|---|---|---|---|---|---|
| AFDEKL | 5.70 | 361.6892 | 2 | 288.60 | 36,673 | 6.08 | 88.2 | 42.5 | 40.9 |
| DAFLGS | 3.91 | 305.1472 | 2 | 274.91 | 15,458 | 6.56 | 99.9 | 36.2 | 41.2 |
| DTHKSE * | 1.19 | 358.6637 | 2 | 292.00 | 4537 | 2.90 | 95.4 | 8.3 | 11.6 |
| KDAIPE | 4.55 | 336.6810 | 2 | 286.40 | 952 | 7.69 | 71.7 | 35.6 | 27.2 |
| KFGERA | 3.66 | 354.1946 | 2 | 298.79 | 1133 | 3.60 | 93.9 | 22.1 | 27.9 |
| KFPKAE | 4.01 | 360.2070 | 2 | 308.56 | 2291 | 5.69 | 100.0 | 39.4 | 44.3 |
| LFGDEL | 7.43 | 693.3452 | 1 | 253.69 | 902 | 10.18 | 100.0 | 34.2 | 32.5 |
| LLPKIE | 6.10 | 356.7331 | 2 | 315.47 | 1720 | 7.65 | 100.0 | 41.7 | 46.1 |
| LPKIET | 5.60 | 350.7149 | 2 | 292.34 | 7905 | 6.27 | 96.9 | 31.0 | 31.1 |
| NLPPLT | 6.57 | 654.3815 | 1 | 248.25 | 1517 | 7.63 | 80.2 | 51.5 | 43.0 |
| TVFDKL | 6.40 | 361.7072 | 2 | 295.17 | 2718 | 5.65 | 100.0 | 35.9 | 40.2 |
| VEGPKL | 5.33 | 321.6941 | 2 | 280.58 | 9864 | 5.33 | 85.6 | 38.7 | 39.6 |
| VSTPTL | 6.14 | 617.3495 | 1 | 238.79 | 674 | 5.56 | 100.0 | 43.6 | 46.0 |
References
- Cruz, S.H.; Cilli, E.M.; Ernandes, J.R. Structural Complexity of the Nitrogen Source and Influence on Yeast Growth and Fer-mentation. J. Inst. Brew. 2002, 108, 54–61. [Google Scholar] [CrossRef]
- Lei, H.; Zhao, H.; Zhao, M. Proteases supplementation to high gravity worts enhances fermentation performance of brewer’s yeast. Biochem. Eng. J. 2013, 77, 1–6. [Google Scholar] [CrossRef]
- Verbelen, P.J.; Delvaux, F.R. Brewing yeast in action: Beer fermentation. Appl. Mycol. 2009, 7, 110–135. [Google Scholar] [CrossRef]
- Nisbet, M.A.; Martinson, T.E.; Mansfield, A.K. Accumulation and Prediction of Yeast Assimilable Nitrogen in New York Winegrape Cultivars. Am. J. Enol. Vitic. 2014, 65, 325–332. [Google Scholar] [CrossRef]
- Gibson, B.R.; Lawrence, S.; LeClaire, J.P.R.; Powell, C.; Smart, K.A. Yeast responses to stresses associated with industrial brewery handling: Figure 1. FEMS Microbiol. Rev. 2007, 31, 535–569. [Google Scholar] [CrossRef] [Green Version]
- Malfeito-Ferreira, M. Yeasts and wine off-flavours: A technological perspective. Ann. Microbiol. 2010, 61, 95–102. [Google Scholar] [CrossRef]
- Vilanova, M.; Ugliano, M.; Varela, C.; Siebert, T.; Pretorius, I.S.; Henschke, P.A. Assimilable Nitrogen Utilisation and Produc-tion of Volatile and Non-Volatile Compounds in Chemically Defined Medium by Saccharomyces Cerevisiae Wine Yeasts. Appl. Microbiol. Biotechnol. 2007, 77, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Bely, M.; Rinaldi, A.; Dubourdieu, D. Influence of Assimilable Nitrogen on Volatile Acidity Production by Saccharomyces Cerevisiae during High Sugar Fermentation. J. Biosci. Bioeng. 2003, 96, 507–512. [Google Scholar] [CrossRef]
- Becerra-Rodríguez, C.; Marsit, S.; Galeote, V. Diversity of Oligopeptide Transport in Yeast and Its Impact on Adaptation to Winemaking Conditions. Front. Genet. 2020, 11, 602. [Google Scholar] [CrossRef]
- Becerra-Rodríguez, C.; Taghouti, G.; Portier, P.; Dequin, S.; Casal, M.; Paiva, S.; Galeote, V. Yeast Plasma Membrane Fungal Oligopeptide Transporters Display Distinct Substrate Preferences despite Their High Sequence Identity. J. Fungi 2021, 7, 963. [Google Scholar] [CrossRef]
- Ganapathy, V.; Leibach, F.H. Proton-coupled solute transport in the animal cell plasma membrane. Curr. Opin. Cell Biol. 1991, 3, 695–701. [Google Scholar] [CrossRef]
- Marsit, S.; Mena, A.; Bigey, F.; Sauvage, F.-X.; Couloux, A.; Guy, J.; Legras, J.-L.; Barrio, E.; Dequin, S.; Galeote, V. Evolutionary Advantage Conferred by an Eukaryote-to-Eukaryote Gene Transfer Event in Wine Yeasts. Mol. Biol. Evol. 2015, 32, 1695–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.W.; Smith, M.W. Peptide Transport by Micro-Organisms. Adv. Microb. Physiol. 1994, 36, 1–80. [Google Scholar] [CrossRef] [PubMed]
- Damon, C.; Vallon, L.; Zimmermann, S.; Haider, M.Z.; Galeote, V.; Dequin, S.; Luis, P.; Fraissinet-Tachet, L.; Marmeisse, R. A novel fungal family of oligopeptide transporters identified by functional metatranscriptomics of soil eukaryotes. ISME J. 2011, 5, 1871–1880. [Google Scholar] [CrossRef]
- Hauser, M.; Donhardt, A.M.; Barnes, D.; Naider, F.; Becker, J.M. Enkephalins Are Transported by a Novel Eukaryotic Peptide Uptake System. J. Biol. Chem. 2000, 275, 3037–3041. [Google Scholar] [CrossRef] [Green Version]
- Lubkowitz, M.A.; Barnes, D.; Breslav, M.; Burchfield, A.; Naider, F.; Becker, J.M. Schizosaccharomyces pombe isp4 encodes a transporter representing a novel family of oligopeptide transporters. Mol. Microbiol. 2002, 28, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Lubkowitz, M.A.; Hauser, L.; Breslav, M.; Naider, F.; Becker, J.M. An oligopeptide transport gene from Candida albicans. Microbiology 1997, 143, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Wiles, A.M.; Cai, H.; Naider, F.; Becker, J.M. Nutrient regulation of oligopeptide transport in Saccharomyces cerevisiae. Microbiology 2006, 152, 3133–3145. [Google Scholar] [CrossRef] [Green Version]
- Harscoat-Schiavo, C.; Nioi, C.; Ronat-Heit, E.; Paris, C.; Vanderesse, R.; Fournier, F.; Marc, I. Hydrophilic properties as a new contribution for computer-aided identification of short peptides in complex mixtures. Anal. Bioanal. Chem. 2012, 403, 1939–1949. [Google Scholar] [CrossRef]
- Huang, Y.-P.; Dias, F.F.G.; de Moura, J.M.L.N.; Barile, D. A complete workflow for discovering small bioactive peptides in foods by LC-MS/MS: A case study on almonds. Food Chem. 2021, 369, 130834. [Google Scholar] [CrossRef]
- Le Maux, S.; Nongonierma, A.B.; Murray, B.; Kelly, P.M.; FitzGerald, R.J. Identification of short peptide sequences in the nanofiltration permeate of a bioactive whey protein hydrolysate. Food Res. Int. 2015, 77, 534–539. [Google Scholar] [CrossRef]
- Piovesana, S.; Capriotti, A.L.; Cerrato, A.; Crescenzi, C.; La Barbera, G.; Laganà, A.; Montone, C.M.; Cavaliere, C. Graphitized Carbon Black Enrichment and UHPLC-MS/MS Allow to Meet the Challenge of Small Chain Peptidomics in Urine. Anal. Chem. 2019, 91, 11474–11481. [Google Scholar] [CrossRef] [PubMed]
- Sonsmann, G.; Römer, A.; Schomburg, D. Investigation of the influence of charge derivatization on the fragmentation of multiply protonated peptides. J. Am. Soc. Mass Spectrom. 2002, 13, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Mesa, M.; Escourrou, A.; Monteau, F.; Le Bizec, B.; Dervilly-Pinel, G. Current applications and perspectives of ion mobility spectrometry to answer chemical food safety issues. TrAC Trends Anal. Chem. 2017, 94, 39–53. [Google Scholar] [CrossRef]
- Arju, G.; Taivosalo, A.; Pismennoi, D.; Lints, T.; Vilu, R.; Daneberga, Z.; Vorslova, S.; Renkonen, R.; Joenvaara, S. Application of the UHPLC-DIA-HRMS Method for Determination of Cheese Peptides. Foods 2020, 9, 979. [Google Scholar] [CrossRef]
- Campuzano, I.D.G.; Giles, K. Historical, Current and Future Developments of Travelling Wave Ion Mobility Mass Spectrom-etry: A Personal Perspective. TrAC Trends Anal. Chem. 2019, 120, 115620. [Google Scholar] [CrossRef]
- Fiechter, G.; Mayer, H. UPLC analysis of free amino acids in wines: Profiling of on-lees aged wines. J. Chromatogr. B 2011, 879, 1361–1366. [Google Scholar] [CrossRef]
- Salmon, J.-M.; Barre, P. Improvement of Nitrogen Assimilation and Fermentation Kinetics under Enological Conditions by Derepression of Alternative Nitrogen-Assimilatory Pathways in an Industrial Saccharomyces cerevisiae Strain. Appl. Environ. Microbiol. 1998, 64, 3831–3837. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.-J.; Henschke, P.A. Implications of Nitrogen Nutrition for Grapes, Fermentation and Wine. Aust. J. Grape Wine Res. 2008, 11, 242–295. [Google Scholar] [CrossRef]
- Bely, M.; Sablayrolles, J.-M.; Barre, P. Automatic detection of assimilable nitrogen deficiencies during alcoholic fermentation in oenological conditions. J. Ferment. Bioeng. 1990, 70, 246–252. [Google Scholar] [CrossRef]
- Adler-Nissen, J. Enzymic Hydrolysis of Food Proteins; Elsevier Science Limited: London, UK, 1986. [Google Scholar]
- Pasupuleti, V.K.; Demain, A.L. Protein Hydrolysates in Biotechnology; Springer: Cham, Switzerland, 2010. [Google Scholar] [CrossRef]
- Aloo, S.O.; Oh, D.-H. The Functional Interplay between Gut Microbiota, Protein Hydrolysates/Bioactive Peptides, and Obesity: A Critical Review on the Study Advances. Antioxidants 2022, 11, 333. [Google Scholar] [CrossRef] [PubMed]
- Colla, G.; Hoagland, L.; Ruzzi, M.; Cardarelli, M.; Bonini, P.; Canaguier, R.; Rouphael, Y. Biostimulant Action of Protein Hy-drolysates: Unraveling Their Effects on Plant Physiology and Microbiome. Front. Plant Sci. 2017, 8, 2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loon, L.J.; Kies, A.K.; Saris, W.H. Protein and protein hydrolysates in sports nutrition. Int. J. Sport Nutr. Exerc. Metab. 2007, 17, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Liigand, P.; Kaupmees, K.; Kruve, A. Influence of the amino acid composition on the ionization efficiencies of small peptides. Biol. Mass Spectrom. 2019, 54, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson-Bunch, C.; Sanford, J.A.; Hansen, J.R.; Gritsenko, M.A.; Rodland, K.D.; Piehowski, P.D.; Qian, W.-J.; Adkins, J.N. Assessment of TMT Labeling Efficiency in Large-Scale Quantitative Proteomics: The Critical Effect of Sample pH. ACS Omega 2021, 6, 12660–12666. [Google Scholar] [CrossRef]
- Waliczek, M.; Kijewska, M.; Rudowska, M.; Setner, B.; Stefanowicz, P.; Szewczuk, Z. Peptides Labeled with Pyridinium Salts for Sensitive Detection and Sequencing by Electrospray Tandem Mass Spectrometry. Sci. Rep. 2016, 6, 37720. [Google Scholar] [CrossRef]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, H.; Clauser, K.; Mertins, P.; Carr, S.A.; Kuster, B. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol. Cell. Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arju, G.; Berg, H.Y.; Lints, T.; Nisamedtinov, I. Methodology for Analysis of Peptide Consumption by Yeast during Fermentation of Enzymatic Protein Hydrolysate Supplemented Synthetic Medium Using UPLC-IMS-HRMS. Fermentation 2022, 8, 145. https://doi.org/10.3390/fermentation8040145
Arju G, Berg HY, Lints T, Nisamedtinov I. Methodology for Analysis of Peptide Consumption by Yeast during Fermentation of Enzymatic Protein Hydrolysate Supplemented Synthetic Medium Using UPLC-IMS-HRMS. Fermentation. 2022; 8(4):145. https://doi.org/10.3390/fermentation8040145
Chicago/Turabian StyleArju, Georg, Hidde Yaël Berg, Taivo Lints, and Ildar Nisamedtinov. 2022. "Methodology for Analysis of Peptide Consumption by Yeast during Fermentation of Enzymatic Protein Hydrolysate Supplemented Synthetic Medium Using UPLC-IMS-HRMS" Fermentation 8, no. 4: 145. https://doi.org/10.3390/fermentation8040145
APA StyleArju, G., Berg, H. Y., Lints, T., & Nisamedtinov, I. (2022). Methodology for Analysis of Peptide Consumption by Yeast during Fermentation of Enzymatic Protein Hydrolysate Supplemented Synthetic Medium Using UPLC-IMS-HRMS. Fermentation, 8(4), 145. https://doi.org/10.3390/fermentation8040145